编者按:
2020 年 12 月 21 日,国内著名英文期刊 Protein & Cell 与热心肠研究院合作,成功举办了“Protein & Cell人类微生物组专刊线上论坛”活动。今天我们特别整理发布来自中国科学院遗传与发育生物学研究所工程师刘永鑫博士的演讲视频及图文实录,希望能助你涨知识。

大家好,我叫刘永鑫,来自中科院遗传发育所,也是《宏基因组》公众号的创始人。
感谢热心肠研究院的邀请,今天为大家分享我最近在 Protein & Cell 发表的一篇综述《微生物组数据分析与可视化实战》。

近 10 年来,关于微生物组的研究也长期霸占着 Cell、Nature、Science 这些世界顶级期刊的封面,得到了科学界极大的关注,也推动了本领域的研究进入了发展的黄金时代。

为什么微生物组的研究这么热呢?
因为人、动物、植物的生活环境中都充满了微生物,而脱离了微生物的生物学研究是不完整的。

以人类自身为例,我们人作为微生物的宿主,人体本身只有 25000 多个编码基因,而人类肠道中已经发现了 1.7 亿个基因、20 多万个基因组、近 5000 个物种。这些数据也不是来自同一个人,是基于目前近十年上万个人的研究的累计结果。典型的个体中也包括 160 多个物种。
人类的基因组是固定的,近年来发现的基因极少,也基本不再更新。而人体肠道菌群中的基因的研究,随着我们探索更多的人群,也随着技术的发展,将会持续地进一步发现更多的基因和基因组。
我们面对如此复杂的基因组,该如何进行研究呢?

今年我也有幸受到了 Protein & Cell 杂志的邀请,撰写了一篇关于微生物组数据分析的综述,分享了一下我近 5 年来在微生物组研究中积累的一些经验,希望让同行少走弯路,高效开展微生物组数据分析,更好地探索微生物组中的生物学规律。此文章上线半年已被引用 9 次。
(综述全文请访问:https://doi.org/10.1007/s13238-020-00724-8)

我总结,开展微生物组研究主要分为三步走。首先是测序方法的选择,其次是对测序数据中的物种和功能进行定量,最后是结果的统计和可视化。

首先是微生物组研究技术。

我总结,基于高通量测序的微生物组研究,主要分为 3 个层面的 5 大类研究。
首先是微生物组层面,我们最重要的研究是开展培养组学研究。培养组学可以获得可培养的微生物,它具有高通量和靶向的特点,但是也受到实验成本高、工作量大,以及选择培养基和环境的限制,目前仍没有广泛的应用。
我们目前研究较多的方法,是基于 DNA 层面,因为 DNA 比较好提取,而且也比较好保存。
因此,在 DNA 层面也开发出了多个技术,如目前最广泛使用的扩增子测序技术,我们通常以 16S、18S 或 ITS 等 marker(标志物)基因为研究对象,采用 PCR 的方法即可扩增出菌群的群落结构。
它具有成本低、通量也比较高的优点,可以开展大规模的研究。扩增子得到的数据量并不大,所以我们在自己的笔记本上就可以开展成百上千个样本的分析。扩增子每年也支撑了上万篇文章的发表。
扩增子测序也有一些局限性,比如 PCR 扩增有一定的偏好性,同时它的精度也只有属的水平,而且只能研究物种的分类,不能研究功能。
想要解决这些不足,我们就要开展宏基因组测序的研究。
宏基因组测序可以达到菌株的水平精度,同时也可以获得这些微生物组的潜在功能,甚至可以进一步挖掘不可培养微生物的基因组草图。但宏基因组也容易出现成本高、宿主污染严重等问题,目前每年只能支撑上千篇文章的发表。不过,它也代表着目前发展最快的主流研究方法。
值得一提的还有宏病毒组研究方法。因为病毒是很特殊的一类生物,既有 DNA 病毒,又有 RNA 病毒,所以开展完整的宏病毒组的研究,需要同时从 DNA 层面的宏基因组测序和 RNA 层面的宏转录组测序做起。例如今年大流行的新冠病毒,它就是一类 RNA 病毒。
最后一个层面是在 mRNA 层面开展的宏转录组测序。宏转录组测序可以获得当前有活性的微生物的现状,也可以研究转录层面微生物组对环境的响应。但是由于 RNA 提取和保存比较困难,测序和分析的成本也比较高,目前并没有得到特别广泛的应用。

关于培养组层面,由于它至关重要,它架起了微生物组相关分析和功能研究的桥梁,为功能研究提供重要的细菌材料基础,所以我们也开发了一套培养组学的实验方法和相应的分析平台。
我们主要是基于梯度稀释的方法,可以快速地获得数万个微生物的培养物,然后采取类似于扩增子测序的方法,采用双端标签的高通量测序方式,可以低成本地鉴定数万个培养物的物种分类,然后可以建立高通量的细菌资源库,为下游的功能实验做准备。
由于很多生物学同行没有很强大的生物信息学背景,我们也开发了一个在线的分析平台,可以把获得的培养组学数据在线提交,可以快速解析其中的微生物组成和获得分析报告,推动大家培养组学的应用。
该方法也于今年被 Nature Protocol 杂志接收,过几周就会上线正式发表,到时我们的公众号也会同步地进行介绍和宣传。
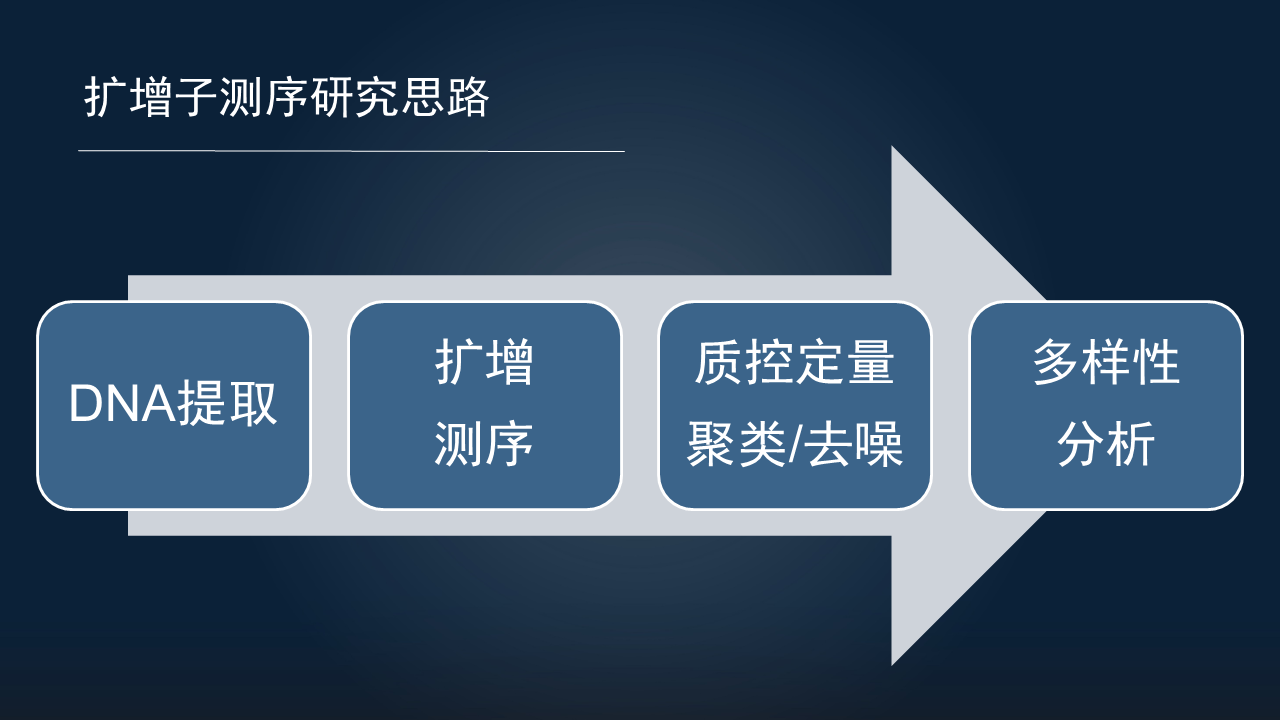
目前使用最广泛的扩增子研究,它的研究思路也主要分为以下 4 步。
首先是研究对象 DNA 的提取;其次是对提取 DNA 的扩增和上机的测序;对获得数据的质量控制和定量,主要分为聚类和去噪两种方式,获得代表性序列;最后是对微生物组的多样性进行研究。
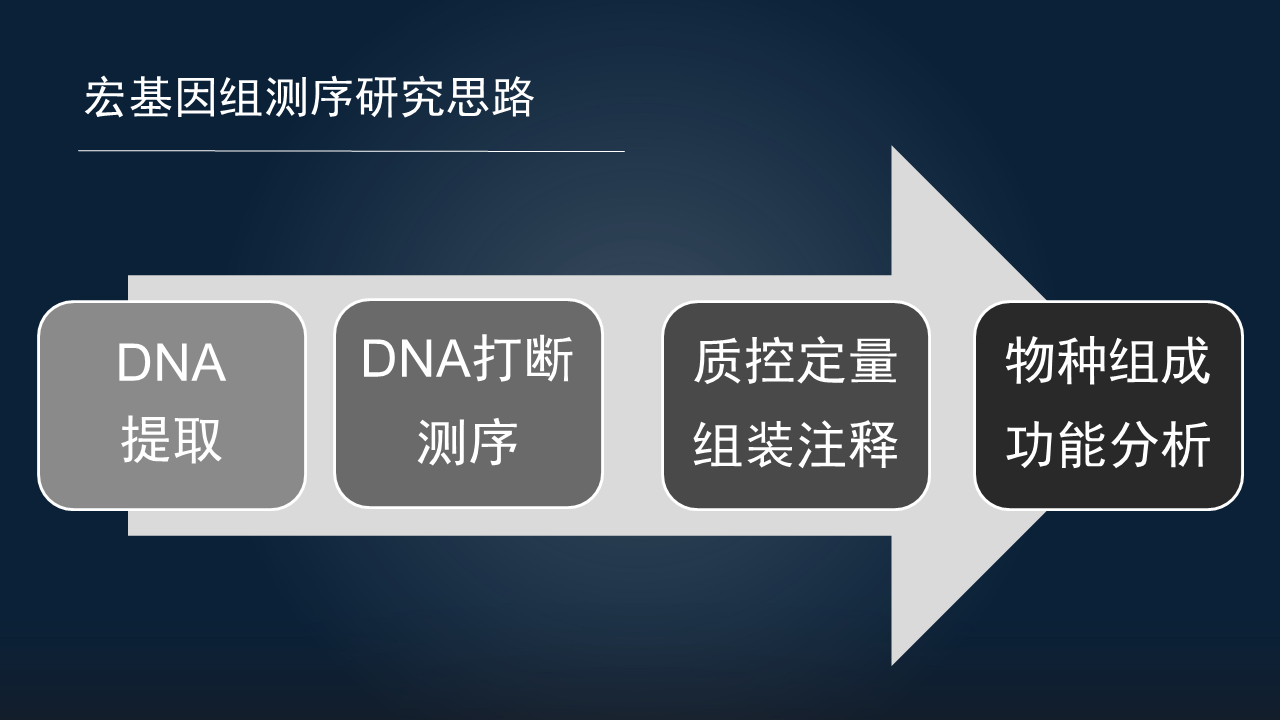
而宏基因组的研究思路也是类似的,也主要分为以下 4 个部分。
对样本 DNA 的提取;由于是短片段测序,我们还需要对样本进行 DNA 的打断,然后再开展测序;结果也是需要质控和定量,对于全新的环境类的样本,我们可能还需要进行组装和注释;下游都是获得物种和功能组成的表,以进行下游分析。

对于微生物组研究实验阶段研究方法相对较缺乏的现状,我们也联合 Bio-protocol 杂志社共同创立了《微生物组实验手册》项目。
我们目前已经邀请到了国内 50 多家单位、100 多个团队的参与,目前正在撰写 200 多篇实验方法的文章,希望给同行提供高质量的实验方法参考,也希望能够统一一些行业标准,为将来的大数据整合分析做准备。
此项目也是长期项目,欢迎有相关研究经验的团队联系我们,发表你的高质量的研究方法,为本领域造福。

当我们获得了微生物组数据之后,第二阶段就需要将数据降维,而在这里,我们主要介绍最广泛使用的扩增子和宏基因组数据的分析流程。
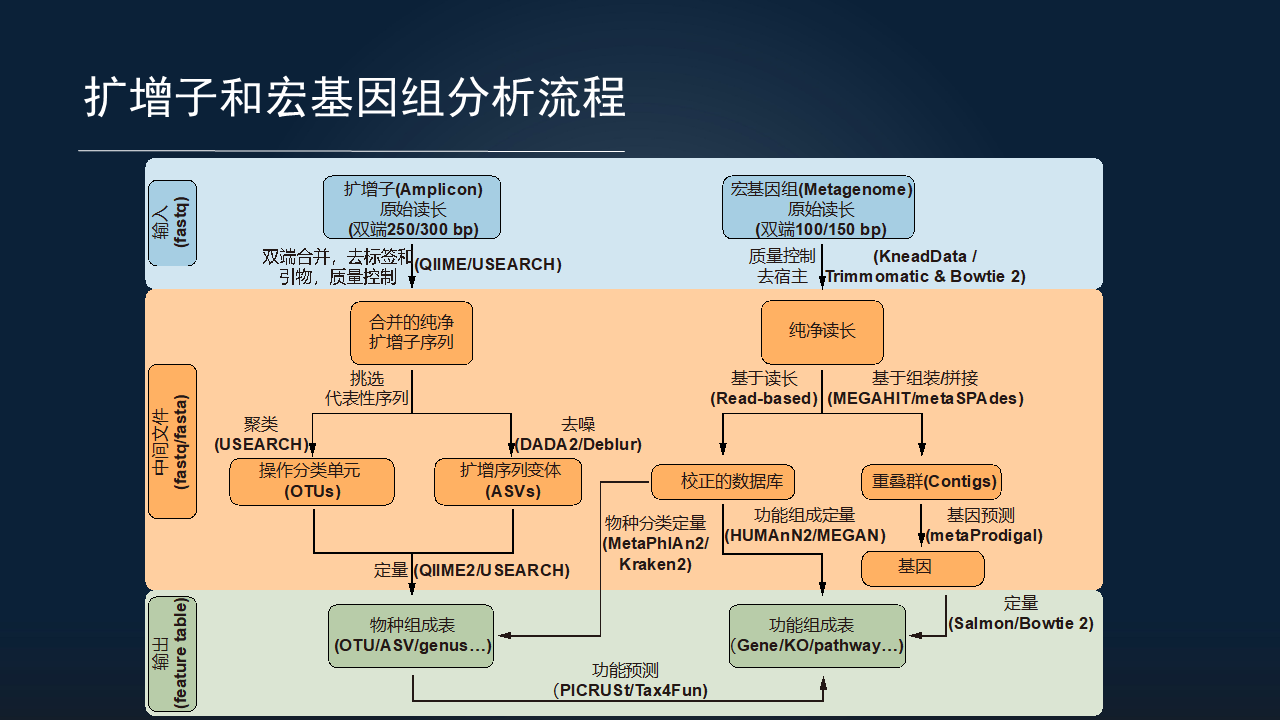
我把扩增子和宏基因组分析流程总结成以下这一张图,主要分为输入数据、中间的处理过程和最终的结果。
无论是扩增子,还是宏基因组,它们的输入数据主要都是 Illumina 的双端数据为主导。主要的区别是它们的读长略有不同,扩增子通常采用 PE250 或 PE300 的模式;而宏基因组通常采用双端 100 或 150 的模式。
它们的结果也是类似的,都包括一个物种组成表和一个功能组成表。
关于中间的过程,扩增子主要有两类的分析方法。一类是基于传统的聚类获得 OTU 的方法,代表的软件有 USEARCH 等;另一类是基于去噪的挑选扩增序列变体的 ASV 的方法,代表的软件也有 DADA2、Deblur。
而宏基因组的分析,目前主流也有两大类分析方法。一类是基于 reads(读长)层面的,也叫做 Read-based 方法,可以把测序的序列直接比对到数据库上,获得物种和功能的组成。这一类比较适合于人类的研究,因为目前人类的研究中,数据库质量比较高。
而对于其他环境来讲,我们通常采用第二种——基于组装的方法,把原始序列组装成重叠群(contig),来进一步挖掘基因,甚至是挖掘潜在的未培养微生物的基因组。

关于微生物组,到底选用哪些软件呢?
在扩增子分析上,我们推荐大家使用目前最高引用的 QIIME 系列,它已经被引用了 2 万多次。而且新发表的 QIIME 2 具有可重复性——全程记录分析过程和图表可交互等特点,你也可以根据自己的需求去定制、拓展一些功能的插件。
我也有幸在近 4 年里一直参与本项目,而且每年还负责本项目数十万字的中文文档的编写和更新工作。
对于新进入本领域的同行,欢迎系统学习一下 QIIME 2 的 10 万字中文文档。
如果你觉得这个文档比较长,学习需要一两周的时间,没有这么多时间,而只是初步的涉猎本领域的研究,也可以学习我们编写的简明教程。这个教程只有 6000 多字,只要花半天的时间,就可以掌握本软件的安装、基本流程和一些常见结果。
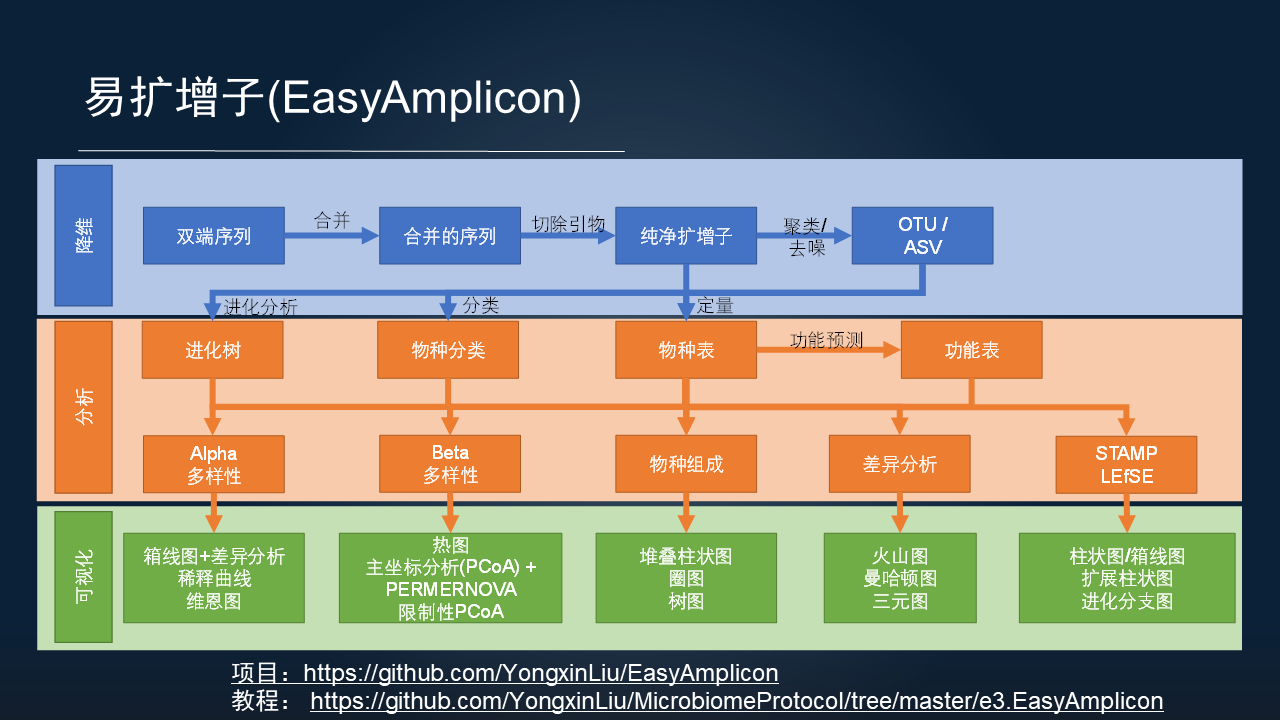
此外,我们也把我们近 5 年对于扩增子方面积累的一些经验和代码,总结成了一套流程,命名为“易扩增子”。它比目前主流的方法在分析速度上更快,也有更多个性化的分析。
如果有需要的话,可以从PPT上的网址访问我们的项目,去安装本套流程,也有配套的中文文档教程。
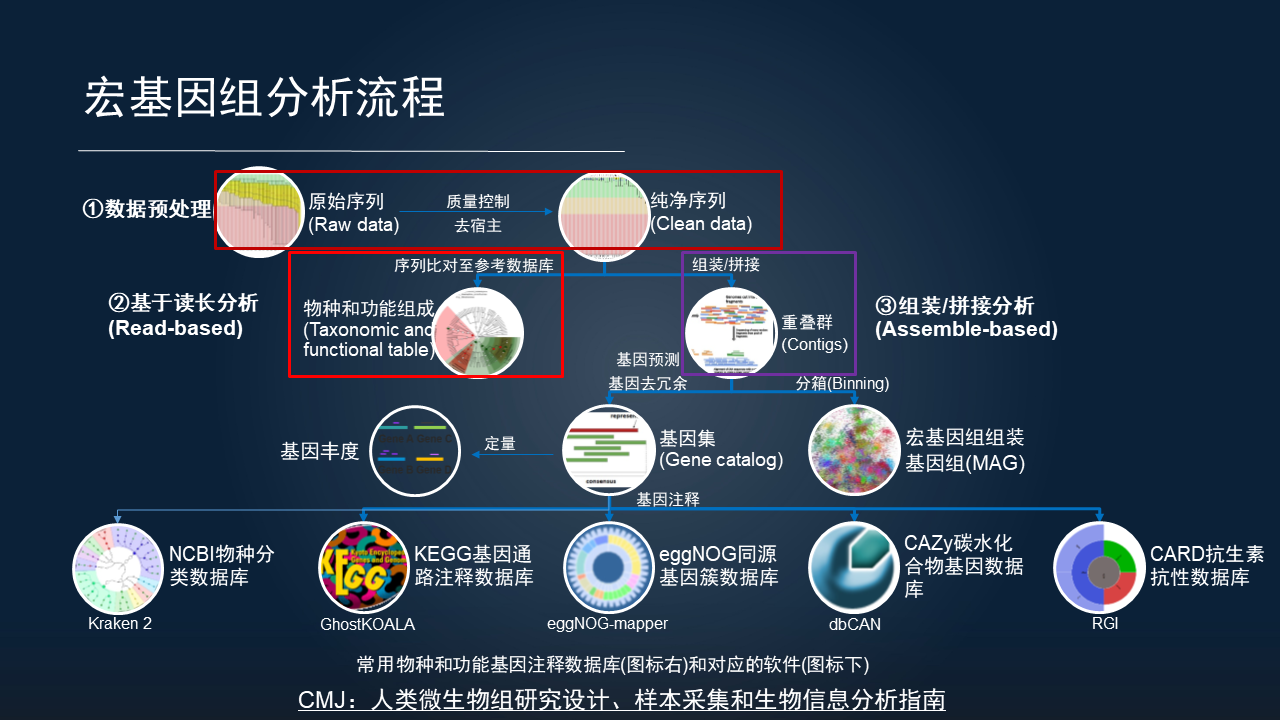
对于宏基因组分析涉及的软件和步骤,就会更复杂一些,而且没有特别成熟的完整分析流程。
我总结,宏基因组分析主要分为三大块。
首先是数据的质量控制和去宿主。我们测序的原始序列不仅要进行质量的控制,而且还要进行宿主的去除。比如人类粪便中宿主还是比较低的,而我们研究的皮肤、口腔,甚至是植物的叶际或根际,都有大量的宿主污染,都达到了 90%,甚至是 99%的含量,所以去除宿主是宏基因组分析中至关重要而又独特的一步。
当我们获得高质量的序列之后,主要分为两类分析。
一类是基于参考数据库的快速的定量分析,也就是 Read-based 分析方法。它比较适合于人类相关的研究,因为在目前已经报道的 20 多万个已知的基因组中,大部分是来自于人类的研究。
如果是开展其他的环境或植物这种非人类的研究,它的参考数据库并不是很完善,所以我们通常采取基于组装的分析,将短序列拼接成重叠群,再进一步的注释基因,根据已有的一些数据库物种和功能的注释来进一步挖掘。
也可以在 contig 的层面上进行进一步的分箱,获得宏基因组组装的基因组,也就是我们目前流行的 MAG 分析,挖掘一些未培养微生物的基因组,去探索细菌基因组中的代谢通路等。

关于宏基因组使用,我们有两个困难,一是宏基因组的软件如何安装,二是这些数据库如何部署。
关于软件的安装,近两年发展出了 Bioconda 体系,可以帮助我们快速地安装生物信息学软件,目前该平台已经收录了上万个生物信息学相关软件,我们可以用简单的命令,快速地安装。下面也有关于我们介绍的 Bioconda 使用的教程,该项目也于 2018 年发表于 Nature Methods 上面。
关于数据库下载,推荐大家从软件配套的官网进行下载,但是在部署中还会遇到一些下载慢的问题,或者是有些数据库是放在国外的 Google 或者是 Dropbox 上面,我们国内是没法下载的。我们也联合微生物所的数据中心,共同建立了国内的微生物组数据库的备份站点,可以方便同行去高速地下载一些微生物组专用分析的数据库。

此外,我们在近 5 年的研究中,也积累了一套比较完善的宏基因组的分析流程,把它命名为“易宏基因组”(EasyMetagenome)。如果大家有需要开展宏基因组分析,而又无从下手的话,可以参考我们在网上共享的分析流程,它也是每个季度实时更新的。
它主要包括 3 个文档。一个是 soft_db,它包括了软件和数据库的安装和部署;一个是 pipeline,包括了常见的质控、去宿主、基于参考数据库的有参分析、基于组装拼接的分析方法和分箱,以及下游基因组的分析等软件的使用和关联的相关脚本。
还有一个 FAQ 文档,是对于我们使用中常见问题的解决方案的描述,这个文档是采用 Markdown 的格式编写的,大家可以在一些 VS Code 或者是在有道云笔记中呈现出目录结构,方便大家浏览和阅读。

最后的部分是,我们对获得的物种或功能组成的特征表进行统计和可视化,以探索潜在的生物学规律。
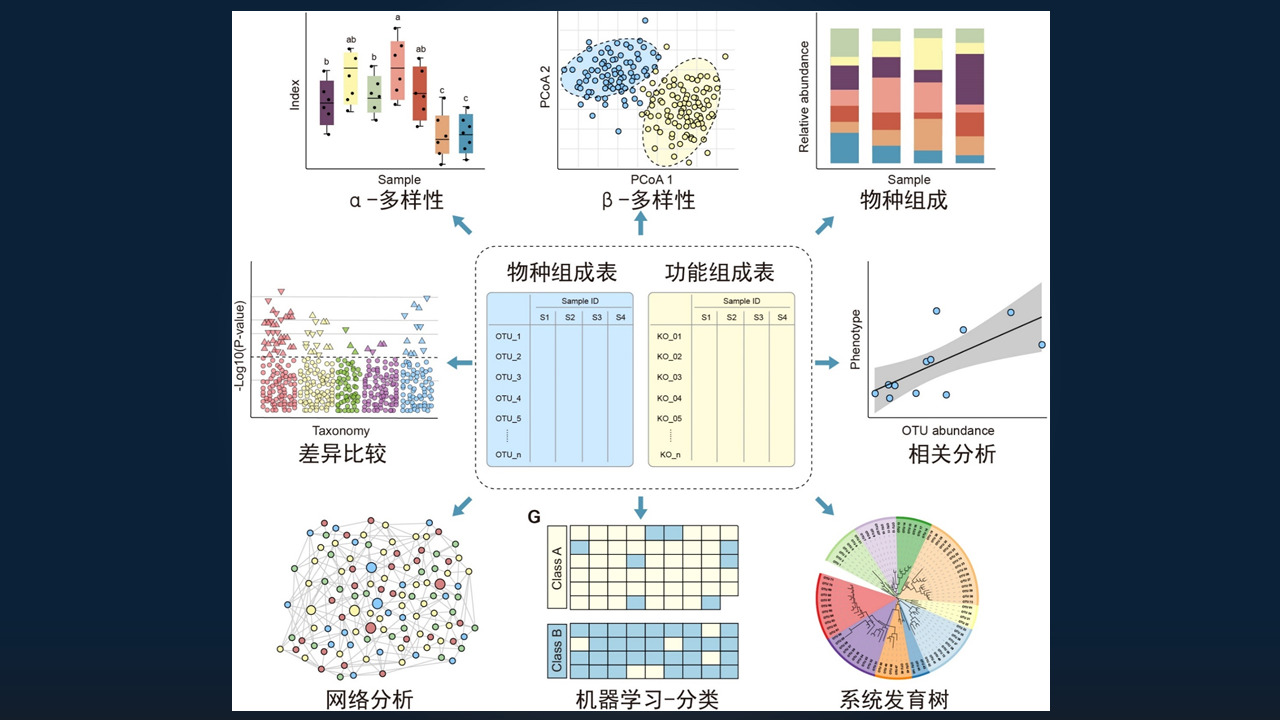
基于物种组成和功能组成的分析,我总结为 8 大类的分析方法,主要包括 α-多样性分析、β-多样性分析、物种组成分析、差异比较、相关分析、网络分析、机器学习和进化分析等。

目前已经有了一些微生物组分析和可视化的R包。例如, vegan 可以提供多样性的分析,phyloseq 可以提供多样性结合进化树的分析,还有今年刚在 Nature Protocol 发表的 MicrobiomeAnalyst,它可以进行在线的,也可以进行本地的双重分析,提供一些高质量的图表。

我们在近 5 年的工作中,也积累了大量分析的代码和一些高质量图表分析的流程,把它们总结成了一个 amplicon 包,大家如果有需要相关图表样式的分析,可以参考我们编写包中的函数,可以一键生成发表级别的样式,欢迎大家使用,也欢迎有经验的同行来贡献你的代码,来扩充我们的代码库,让同行们更方便地进行数据的探索和可视化。
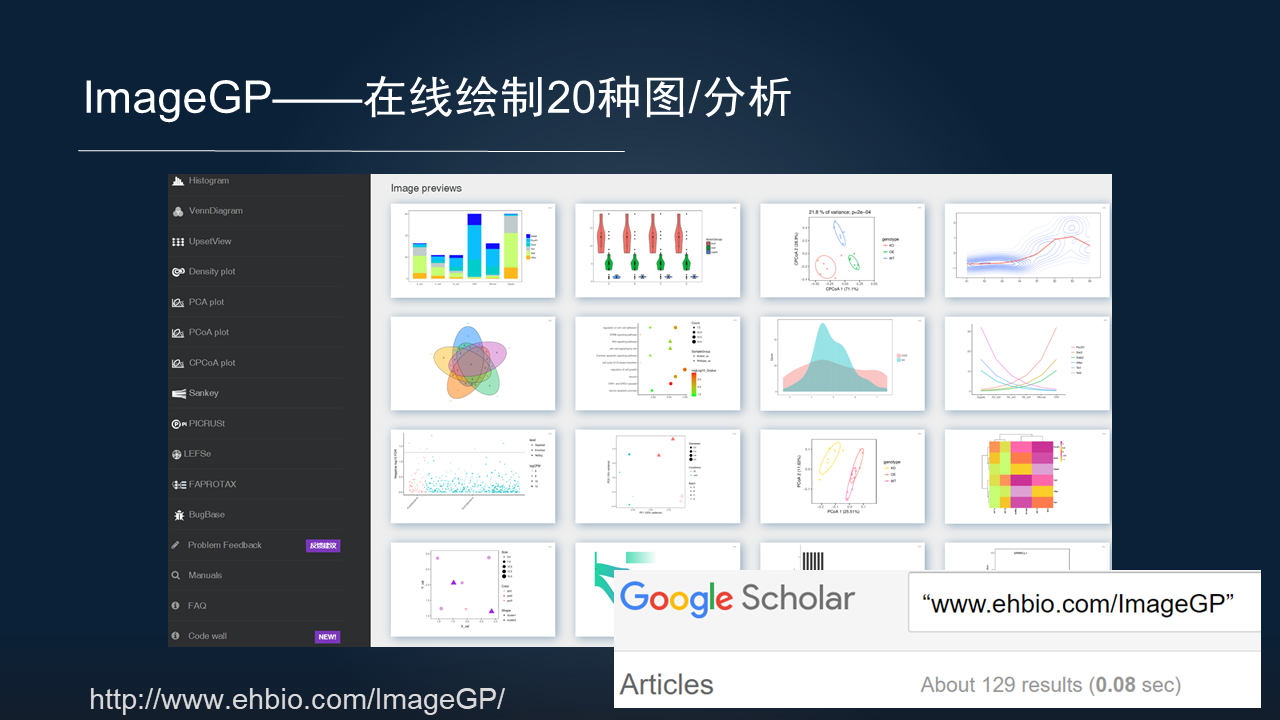
对于很多的生物学同行,是很难在代码层面开展分析,因此,我们也开发了在线分析的网站。
ImageGP 就是我们开发的一个可以在线绘制 20 多种图和分析的一个平台,大家可以访问,把数据粘贴上,一键可以生成发表级别的图表。该平台上线两年,已经被引用了 129 次。
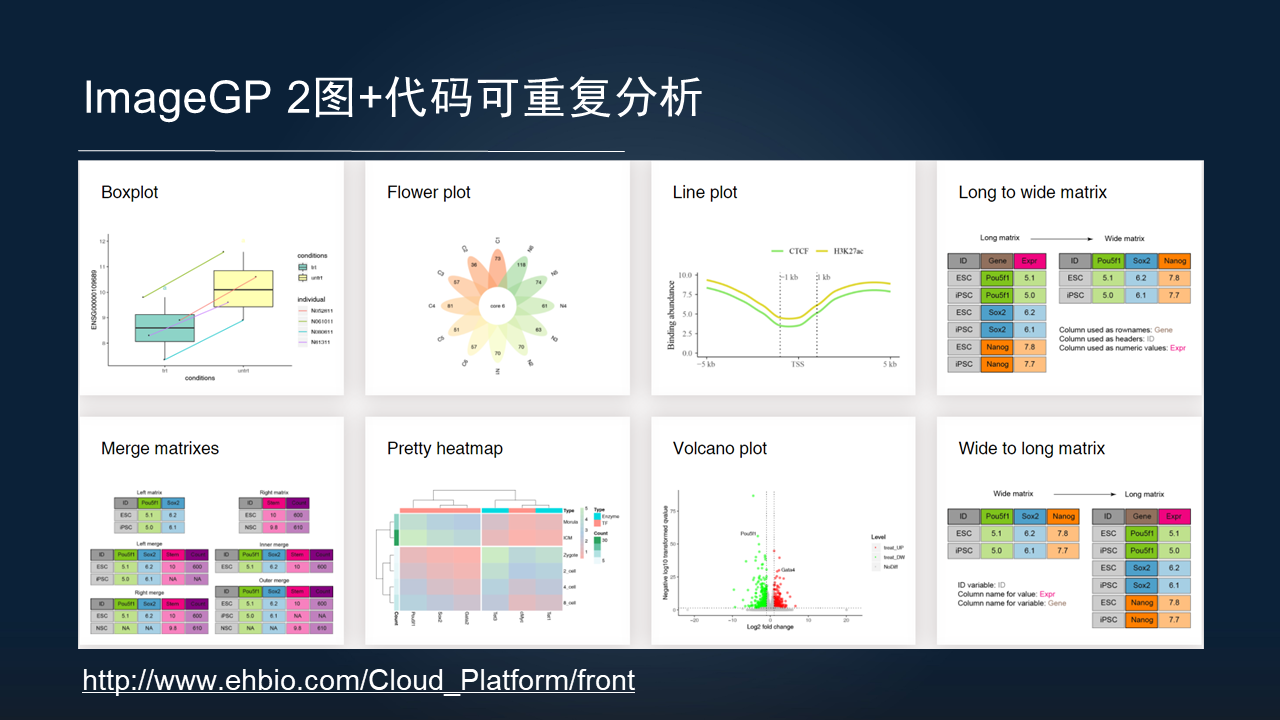
由于目前的分析对可重复的要求越来越高,我们也开发了 Image GP 第 2 版。
这个网站不仅能提供绘图的结果,而且也能对绘图的全过程的代码进行打包下载,这样方便读者,既可以进一步的基于代码修改图片的细节,同时也可以连同代码数据和最后绘制的图表,随着文章一同发表,实现高质量的、可重复分析的标准。

关于微生物组数据分析,它是一门系统的学科,以上这些分析的代码和网站还是远远不够的。
我们目前也正在联合科学出版社,在编写一本《微生物组数据分析》专著,希望能够填补微生物组领域数据分析和可视化资料的空白,提供易学易用的参考书。
我们采用众筹编写的模式,任何感兴趣的同行都可以撰写你比较擅长的分析方法,来提交我们,共同发表。
我们内容格式要求是有背景介绍、实例解读和实战代码三部分,能够帮助同行看懂文章,会归纳总结和写文章,实现自主分析。
我们也计划在一年内完善这个中文专著,并建立起中文的百科平台。对于优质内容,我们也推荐同行发表中英文相应的方法期刊,也希望在两年内建立我国自主的、有影响力的微生物组分析平台,三年能够走出去,发表有国际影响力的方法和外文专著。
该项目正在进展中,目前已经更新了 30 多个章节,大家可以访问下面的链接(点此查看),进一步阅读。也可以在我们公众号获得全部的资源。

总结一下,微生物组研究主要分为三步走,包括研究方法的选择、分析流程的搭建和使用,以及结果的统计和可视化。
关于研究方法,主要包括培养组学、扩增子、宏基因组、宏转录组等。培养组是关键,是大家探索功能的未来;而扩增子,它特别便宜,适合于快速入门,然后结合宏基因组,进一步探索研究对象的功能。
在分析流程上,扩增子分析首选 QIIME 2 分析平台,也可选我们开发的易扩增子,开展更灵活一些的个性化的分析;宏基因组的软件安装,我推荐使用 BioConda;数据库的下载,可以备选我们和中科院微生物所共同建立的数据中心;常用软件的使用代码可以参考我们分享的易宏基因组(EasyMetagenome)的流程。
关于统计和可视化,基于物种和功能组成表的分析,可以开展 α-多样性、β-多样性、物种组成、差异比较、相关、网络、机器学习、进化、来源追溯等十余类的分析。
最后一点,建议大家,任何分析方法只是一个工具,结果的质量更多来自于研究人员专业地选择和判断。更多的信息可以阅读我们撰写了这篇综述(点此查看)。

此外,以上综述内容是远远不够的,我们还成立了《宏基因组》公众号。目前已经三年多,每天保持更新,已经分享了 2400 多篇原创的文章,也有国内 100 多位同行的参与,目前已经达到了近 11 万的同行关注,感兴趣和想进一步学习的同行,可以关注我们的公众号。
此外,我们还有同行的技术交流群,大家可以加我的微信,备注姓名、单位、研究方向,可以加入同行群,进行技术的交流。
最后致谢一下 Protein & Cell 杂志的邀请,才有了这篇综述的问世。
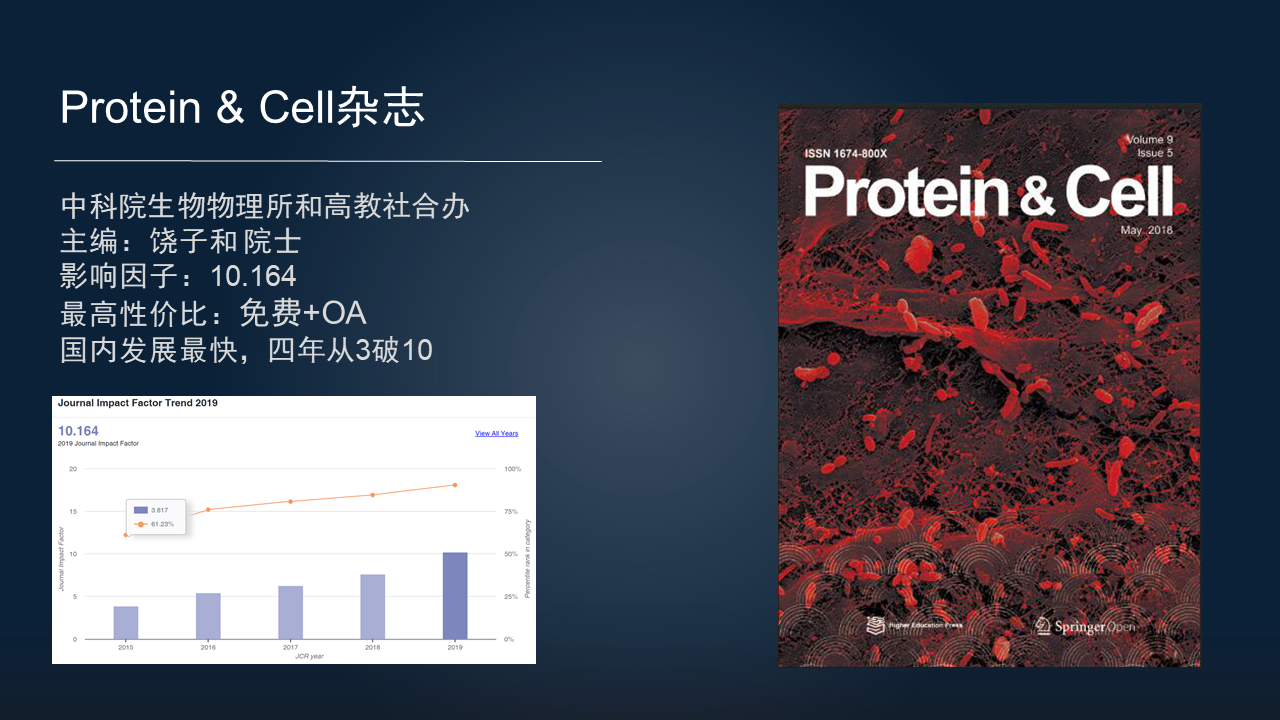
在这里介绍一下 Protein & Cell 杂志,它是中科院生物物理所和高教社合办的一本高质量期刊,主编是饶子和院士,影响因子为 10.164,是我目前见到的最高性价比的杂志,它不仅投稿至发表没有任何费用,而且文章也是 OA 模式,全球任何地方都可以完全免费访问。
而且它在国内也是发展最快速的杂志。4 年来,影响因子从 3 分直接到了破 10,而且还会持续继续发展下去,欢迎大家把优秀的稿件投此期刊,也多读、多引用本杂志的文章。
祝大家开展微生物组学数据分析顺利,发现更多的生物学规律。
感谢大家的聆听!







