编者按:
本文是中国科学院遗传与发育生物学研究所工程师刘永鑫博士发表于 Protein & Cell 期刊上的综述译稿,转载自公众号《宏基因组》。
以下是正文:

文章发表仅半年,Google 学术统计被引 11 次。

扩增子和宏基因组数据分析实用指南
A practical guide to amplicon and metagenomic analysis of microbiome data
Protein Cell——[IF: 10.164]
DOI: https://doi.org/10.1007/s13238-020-00724-8
Review: 2020-5-11
第一作者:刘永鑫1,2,3, 秦媛1,2,3,4, 陈同5
通讯作者:刘永鑫(yxliu@genetics.ac.cn)1,2,3, 白洋(ybai@genetics.ac.cn)1,2,3,4
其他作者:卢美萍6, 钱旭波6, 郭晓璇1,2,3
作者单位:
1.中国科学院遗传与发育生物学研究所,植物基因组学国家重点实验室
2.中国科学院大学,生物互作卓越创新中心
3.中国科学院遗传与发育生物学研究所,中国科学院-英国约翰英纳斯中心植物和微生物科学联合研究中心
4.中国科学院大学,现代农学院
5.中国中医科学院中药资源中心
6.浙江大学医学院附属儿童医院风湿、免疫和变态反应科
摘要
近年来高通量测序技术的发展促进了一系列适合微生物组研究的技术发展,同时也积累了海量数据。然而,微生物组数据分析过程复杂、分析工具种类较多,这限制了广大研究者进入该领域。
本文系统概述了微生物组常用测序技术——扩增子和宏基因组等方法的优缺点,推荐了常用软件、分析流程和数据库,以便研究者选择恰当的分析工具和方法。
此外,我们还介绍了微生物组下游分析通用的统计和可视化方法,包括多样性分析、物种组成分析、差异分析、相关分析、网络分析、机器学习、进化分析、来源追溯以及常用可视化方法。最后我们还介绍了可重复分析方法。
我们希望研究者通过阅读此文能学会分析工具的选择方法和高效的数据分析方法,进而有效地挖掘数据背后的生物学意义。
关键字:
微生物组、宏基因组、标记基因、高通量测序、分析方法、分析流程、可重复性、可视化
1. 概述
微生物组是指整个微生境,包括微生物、基因组和周围环境,定义详见Microbiome:微生物组名词定义。随着高通量测序(high-throughput sequencing, HTS)技术和数据分析方法的发展,近年来微生物组在人类、动物、植物和环境中所起的作用变得越来越清晰,植物中进展可阅读我 2019 年在 Current Opinion 上发表的综述,详见COM:重组菌群体系在根系微生物组研究中应用。这些研究成果彻底改变了我们对微生物组的理解。许多国家已经成功启动了国际微生物组研究计划,例如 NIH 人类微生物组计划(HMP)、人类肠道宏基因组学计划(MetaHIT,Nature:基于宏基因组测序构建人类肠道微生物组参考基因集)、整合 HMP(iHMP10 篇 Nature 专题报导人类微生物组计划2(iHMP)成果及展望)和中国科学院微生物组计划(CAS-CMI,NAR:gcMeta——全球微生物组数据存储和标准化分析平台)。这些项目都取得了令人瞩目的成就,将微生物组研究推向了黄金时代。
扩增子和宏基因组学分析的框架是在近十年建立的。但是,微生物组分析方法及标准在近年来有着迅猛地发展。例如,有人提出了在扩增子数据分析中用扩增子序列变异体(amplicon sequence variants, ASV)代替操作分类单位(operational taxonomic units, OTU)的建议,详见扩增子分析还聚OTU就真OUT了。又如,最近发布了下一代微生物组分析软件 QIIME 2,它是一种可重复、交互式、高效、有社区支持的分析平台,文章解读和教程合集见NBT:QIIME 2可重复、交互式的微生物组分析平台。此外,最近也提出了许多新的方法用于物种分类、机器学习和多组学整合分析,如Nature Protocols:整合宏基因组、代谢组和表型分析的的计算框架。
高通量测序和分析方法的发展为深入了解微生物组的结构和功能提供了新的手段。但是,这些新的发展让研究人员,特别是那些没有生物信息学背景的研究人员在选择合适的软件和分析流程时中变得颇为费劲。为了让研究者了解本领域最新进展,并让大家快速掌握相应的软件选择和使用技巧,我们特推出此综述。在这篇文章中,我们将讨论广泛用于微生物组分析的软件包,总结他们的优点和局限性,并提供了示例代码和选择使用这些工具的窍门。
2. 高通量测序方法介绍

图 1 各种高通量测序方法的优势和局限性:A. 介绍了用于不同分析层次的测序方法。在分子水平上,微生物组研究分为三种层面:微生物、DNA 和 mRNA。相应的研究技术包括培养组、扩增子、宏基因组、宏病毒组和宏转录组。B. 用于微生物组研究测序方法的优缺点。
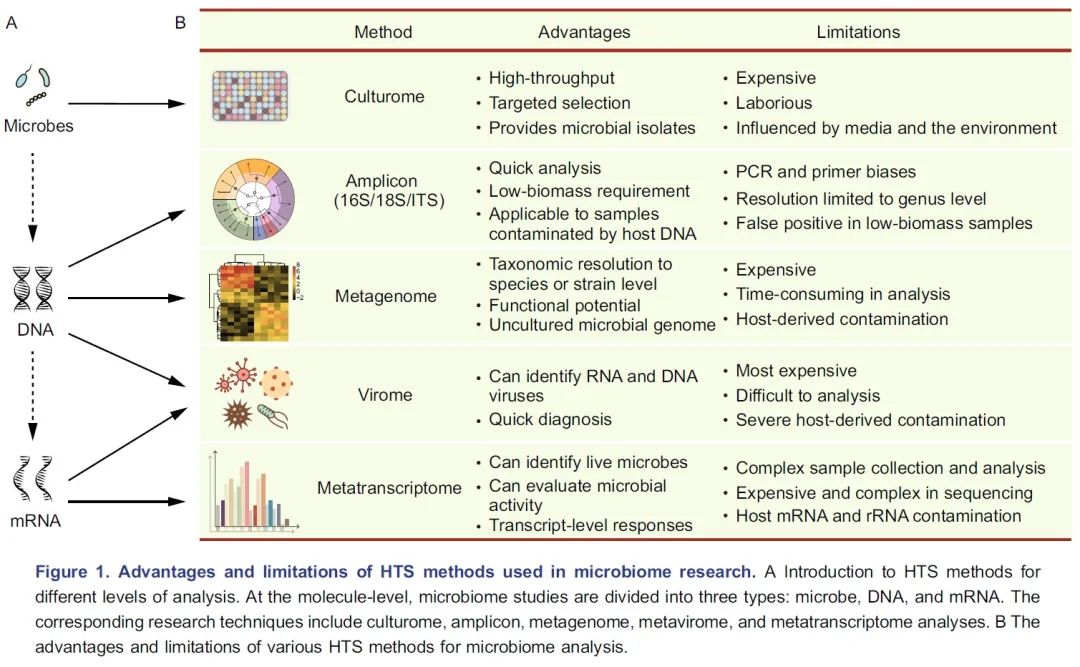
微生物组研究的第一步是了解高通量测序方法具体的优点和局限性。这些测序方法主要用于三个层面的分析:微生物、DNA和mRNA层面(图1A),研究者应根据样本类型和研究目标选择适合的方法。
培养组学(Culturome)是一种在微生物层面培养和鉴定微生物的高通量方法(图1A)。通常微生物的获取方法如下。首先,将样品破碎,根据经验在液体培养基中将其稀释,然后分散至96孔细胞培养板或培养皿中。第二步,将培养板在室温下培养20天。第三步,对每个孔中的微生物进行扩增子测序,并选择纯度高、非冗余菌落孔中的样本作为候选样本。第四步,纯化候选样本并进行16S rDNA全长Sanger测序。最后保留纯化的分离株,详见NBT封面:水稻NRT1.1B基因调控根系微生物组参与氮利用中方法和附图的培养组学流程。培养组学是获得细菌种群最有效的方法,但它昂贵且劳动强度大(图1B)。这种方法在人类、小鼠、海洋沉积物、拟南芥和水稻的微生物组研究中得到开展。这些研究不仅进一步完善了宏基因组学分析的物种分类和功能基因的数据库,而且还为开展功能实验验证提供了细菌基础材料。如想了解培养组学更多的信息请参阅这两篇文献(Lagier et al., 2018 NRM:人类微生物培养及培养组学; Liu et al., 2019 COM:重组菌群体系在根系微生物组研究中应用)。
DNA易于提取、保存和测序,这使研究人员能够开发各种高通量测序的方法(图1A)。微生物组最常用的高通量测序方法是扩增子和宏基因组测序(图1B)。扩增子测序是微生物组分析中使用最广泛的测序方法,几乎可以应用于所有类型的样品。扩增子测序中使用的主要标记基因包括用于原核生物的16S rDNA、用于真核生物的18S rDNA和内转录间隔区(internal transcribed spacers, ITS)。16S rDNA扩增子测序是最常用的方法,但是目前可用引物列表较为混乱。选择引物的一个好方法是先评估其特异性和总体覆盖度,这个过程要么利用真实样品进行,也可以基于SILVA数据库和宿主因素(包括叶绿体、线粒体、核糖体和其他非特异性扩增的潜在来源)进行电子PCR。另一个替代方案是研究者参考与自己研究相似的已发表论文中使用的引物,这样可以节省优化方法的时间,也便于研究结果之间的比较(为整合分析做准备)。两步PCR法通常用于扩增子文库制备,扩增同时向每个样品添加标签(barcode)和接头序列。样品测序通常在Illumina MiSeq、HiSeq 2500或NovaSeq 6000平台上进行,产出双端250bp(PE250)的序列,每个样品含有5~10万条序列。扩增子测序可应用于低生物含量标本或被宿主DNA污染的样品。然而,该技术只能达到“属”级分辨率(主要是由于测序片段较短,通常仅300~500 bp)。此外,它的可靠性受引物和PCR循环次数影响,这可能导致下游分析出现假阳性或假阴性结果(图1B)。
宏基因组测序比扩增子测序提供了更多的信息,但是这种技术较昂贵。对于人类粪便等“纯”样本,每个样本可接受的测序数据量从6~9 GB不等。文库构建和测序的相应价格在100~300美元之间(700~2000元,测序量和耗材纯净级别对价格影响很大)。对于包含复杂微生物群(如土壤)或受宿主DNA污染的样品,每个样本所需的测序量需要30~300 GB。总之,16S rDNA扩增子测序可以用于研究细菌和/或古菌的组成,如果需要更高的物种分类学分辨率和功能信息,则可随机抽取部分样本进行宏基因组测序。当然,假设有足够的可用资金,宏基因组测序可直接用于样本量较小的研究。
宏转录组测序可以分析微生物组中的mRNA,量化基因表达水平,并可提供微生物群落的功能信息。值得注意的是,为了获得微生物组的转录信息,需要有效去除宿主RNA和所有种类的rRNA(图1B)。详见BBI:Eran Elinav组综述在微生物组研究中使用宏转录组。
由于病毒有DNA或RNA作为其遗传物质,从技术上讲,宏病毒组研究包含宏基因组和宏转录组(图1A/B)。由于样品中病毒的生物含量较低,因此要获得足够数量的病毒DNA或RNA进行分析,必须进行病毒富集或去除宿主DNA/RNA(图1B),如NBT:宏基因组中设计全面可扩展探针捕获序列多样性。
测序方法的选择取决于科学问题和样本类型。整合使用不同的方法是很多学者推荐的,因为多组学方法可同时获得微生物组分类和功能的信息。实际上,由于时间和成本的限制,大多数研究人员只能选择一种或两种测序方法。尽管扩增子测序只能提供微生物群的物种分类学组成信息,但它具有成本效益优势(每个样品仅需150~350元),可用于大规模研究。此外,扩增子测序产生的数据量相对较小,分析快速且容易进行。例如,使用普通的便携式计算机在一天之内就可以完成数百个扩增子样品的数据分析。因此,扩增子测序通常用于探索性研究。与扩增子测序相反,宏基因组测序不仅将分类学分辨率扩展到“种”或“株”的水平,而且还提供了潜在的功能信息。宏基因组测序也使得从短片段组装微生物基因组成为可能。但是,对于低生物含量或被宿主基因组严重污染的样品,宏基因组测序并非目前的最佳选择(图1B)。
3. 分析流程
“分析流程”指的是特定程序或脚本,该程序或脚本以一定顺序整合了数个甚至数十个软件程序,用来完成复杂的分析任务。截至2020年6月23日,在Google学术中提及“amplicon”和“metagenome”分别超过24万和4万次。由于他们的广泛使用,我们将讨论用于扩增子和宏基因组分析的当前最佳流程。研究人员应该熟悉Shell环境和R语言,这在我们之前的综述中曾经讨论过,详见遗传:微生物组数据分析方法与应用。
3.1 扩增子分析
扩增子分析的第一阶段任务是将原始序列(一般是fastq格式)转换为特征表。原始序列通常以双端250 bp(PE250)形式从Illumina测序平台生成。本文未讨论如Ion Torrent,PacBio和Nanopore等测序平台,他们产生的数据可能不适合下面讨论的分析流程。分析时,首先将原始序列根据标签(barcode)进行分组,这个过程又叫“拆分”(demultiplexing)。然后将序列合并以获得扩增子序列,并去除标签和引物。通常还需要质量控制步骤以去除低质量的扩增子序列。所有这些步骤都可以使用USEARCH或QIIME完成,或者也可选择测序服务公司提供的纯净扩增子数据用于下一步分析(图2A)。
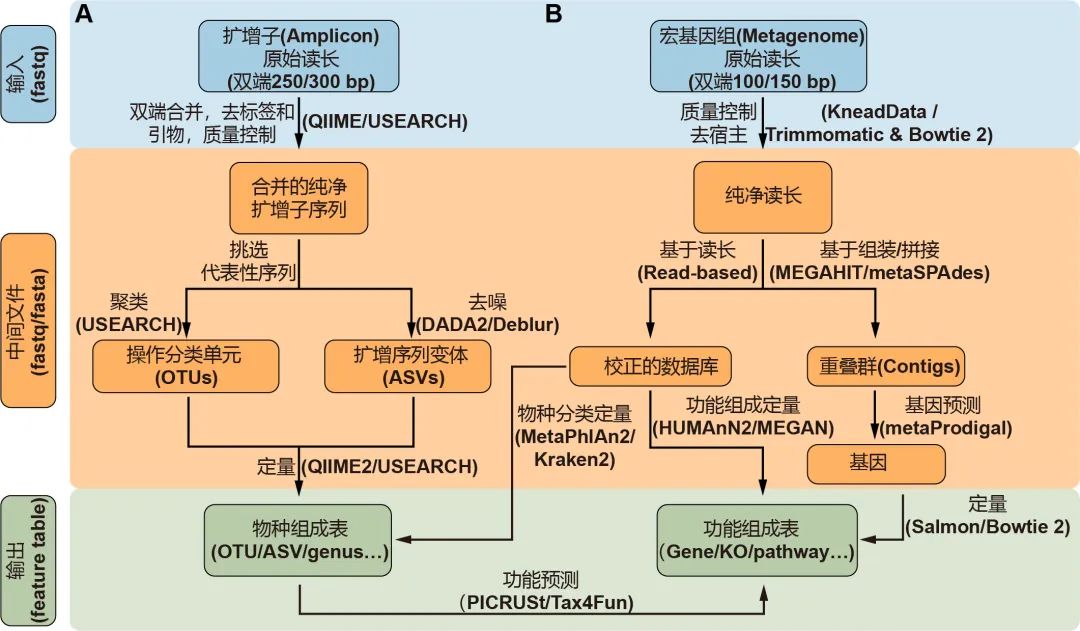
图2. 扩增子(A)和宏基因组(B)测序常用流程:蓝色、橙色和绿色分别代表输入、中间及输出文件。箭头边上的文字代表方法,括号中的是常用软件。物种和功能表格统称为特征表。关于这方面更详细的信息请阅读表1。
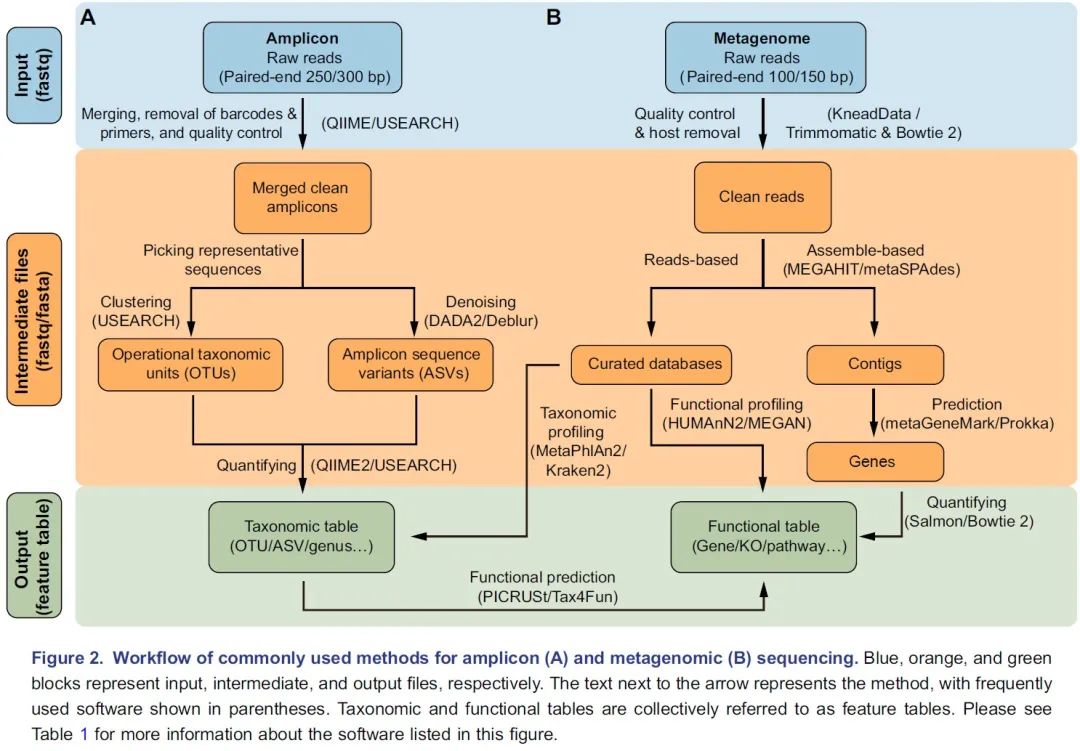
挑选出代表性序列作为物种的代表是扩增子分析的关键步骤,主要包括聚类生成OTU和去噪生成ASV两类方法。UPARSE算法将具有97%相似性的序列聚类为OTU,但此方法可能无法检测“种”或“株”之间的细微差异。DADA2是最近开发的一种去噪算法,可挑选出更准确的代表性序列——ASV。QIIME 2流程中有二种去噪方法可选,即DADA2插件的denoise-paired/single和Deblur插件的denoise-16S,此外USEARCH中的-unoise3也可用于高速去噪并挑选ASV。最后,可以通过量化每个样本中特征序列的频率来获得特征表,即OTU或ASV表。同时,可以对特征序列进行分类,通常在界,门,纲,目,科,属和种的层级上进行分类,从而为微生物群提供了更多级别的降维视角。
通常,16S rDNA扩增子测序只能用于获得有关物种分类组成的信息。但是近几年开发了许多可用的软件包来预测潜在的功能信息。该预测的原理是将16S rDNA序列或分类学信息与数据库中的基因组或文献中的功能描述联系起来。PICRUSt是一种基于Greengenes数据库OTU表的功能预测软件,可用于预测如KEGG通路的宏基因组功能组成信息。新开发的PICRUSt2软件包(https://github.com/picrust/picrust2 )可以基于任意OTU / ASV表直接预测宏基因组功能,详见,NBT:PICRUSt2预测宏基因组功能,PICRUSt2:OTU/ASV等16S序列随意预测宏基因组,参考数据库增大10倍。R包Tax4Fun可以基于SILVA数据库预测微生物群的KEGG功能,Tax4Fun2介绍 。原核生物分类功能注释(FAPROTAX)流程基于已发表微生物的代谢和生态功能执行功能注释 ,例如硝酸盐呼吸、铁呼吸、植物病原体、动物寄生虫或共生体,从而使其可用于对环境、农业和动物微生物组的功能分类和研究。BugBase是Greengenes的扩展数据库,用于预测表型,例如需氧性、革兰氏染色和致病性,该数据库常用于医学研究。
3.2 宏基因组分析
宏基因组测序数据比扩增子测序提供了更高精度的物种组成信息,同时还能提供功能基因的信息,但数据量大、分析过程涉及软件众多,而且一般只能在高性能Linux服务器上开展分析。宏基因组相关软件安装推荐使用Bioconda频道,可有高效安装所需的软件和流程,并自动化解决依赖关系。宏基因组分析计算量大,多任务并行需要队列管理软件防止拥挤,如GNU Parallel软件。
Illumina HiSeqX/NovaSeq系统产出原始数据一般是PE150数据,华大BGI Seq500产生的为PE100数据。宏基因组数据分析的第一个关键的步骤是质量控制和去除宿主污染,这些步骤需要KneadData流程(https://bitbucket.org/biobakery/kneaddata)或联合使用Trimmomatic和Bowtie 2。Trimmomatic是一种灵活的质量控制软件包,适用于Illumina测序数据,可进行修剪低质量序列、文库引物和接头序列。使用Bowtie 2软件,那些与宿主基因匹配的序列将被滤除。KneadData是一个集成的分析流程,包括Trimmomatic、Bowtie 2和相关脚本,可用于质量控制,滤除宿主来源的序列,并输出纯净序列(图2B)。
宏基因组学分析的主要步骤是使用基于序列和/或基于组装的方法将纯净数据转换为物种组成表和功能组成表。基于序列的方法是直接比对纯净序列至预定义的参考数据库,可直接获得特征表(图2B)。MetaPhlAn2是一种常用的生物物种分类学分析工具,可将宏基因序列与预定义的标记基因数据库比对,从而进行生物分类。Kraken 2是基于精确k-mer匹配方法将序列与NCBI中非冗余序列数据库进行匹配,利用最低共同祖先(lowest common ancestor, LCA)算法进行物种分类。分类学的软件众较,其优缺点和适用范围详见2019年Cell发表的关于20种分类软件评测综述(Ye et al., 2019)。HUMAnN2是一种广泛使用的功能定量分析软件,特色是可以用于探索样本内和样本间的贡献多样性(物种对特定功能的贡献)。MEGAN是一种跨平台的图形用户界面(GUI)软件,可进行分类和功能分析(表1)。此外,一些研究提供了特定研究对象的宏基因组参考集,他们可以实现更高的数据利用率和更高质量的物种和功能注释,例如人类肠道2.0(NBT:人类微生物组千万基因的参考基因集)、小鼠肠道(Xiao et al., 2015)、鸡肠道(Microbiome:鸡肠道微生物宏基因集(张和平、魏泓、秦楠点评)*)、海洋OM-RGC.v2(Cell:Tara2.0基因表达的改变和群落的更替塑造了全球海洋宏转录组)、柑橘根际(NC:全球柑橘根际微生物组的结构和功能)等。
基于组装的方法使用MEGAHIT或metaSPAdes等工具将纯净序列组装为长序列,即重叠群(contigs)(图2B)。MEGAHIT用于快速装配大量、复杂的宏基因组数据集,而且内存占用小;而metaSPAdes通常可以生成更长的重叠群,但需要更多的计算资源,详见组装拼接MEGAHIT(多快好省)和评估quast。metaGeneMark或Prokka等软件可以识别出重叠群中的基因。已经装配好的重叠群中的冗余基因需要用CD-HIT等工具去除。最后,可以使用基于比对的工具Bowtie 2或非比对的方法Salmon来生成基因丰度表。宏基因组数据集中基因数量通常在百万级别,需要结合蛋白数据库的层级功能注释实现降维,如KEGG中的KO、模块或通路表。
此外,宏基因组学数据可用于挖掘基因簇或组装微生物基因组草图。AntiSMASH软件和数据库可以挖掘重叠群中潜在的生物合成基因簇,为挖掘新功能基因、酶、代谢通路、代谢物和抗生素等提供了非常重要的线索。分箱(binning)是一种恢复宏基因组数据中部分或完整细菌基因组的方法。可用的分箱工具包括CONCOCT、MaxBin 2和MetaBAT2。分箱工具根据四核苷酸频率和丰度将重重叠群可分类为不同的“箱”(bins),类似于基因组草图。用多个软件优化分析结果和用重新组装的方法可以获得更好的“箱”。我们建议使用MetaWRAP(Microbiome:宏基因组分箱流程MetaWRAP 简介、 安装和数据库部署、 实战和结果解读)或DAStool分箱流程,他们集成了多个分箱工具,可以获得更优的分箱结果、更少的污染,更完整的基因组。这些流程还提供了有用的脚本,用于对箱进行评估和可视化。如想了解宏基因组实验和分析更全面的知识,我们推荐阅读这篇在Nature Biotechnology杂志发表的文献(Quince et al., 2017)。- Nature综述:2万字带你系统入门鸟枪法宏基因组实验和分析。
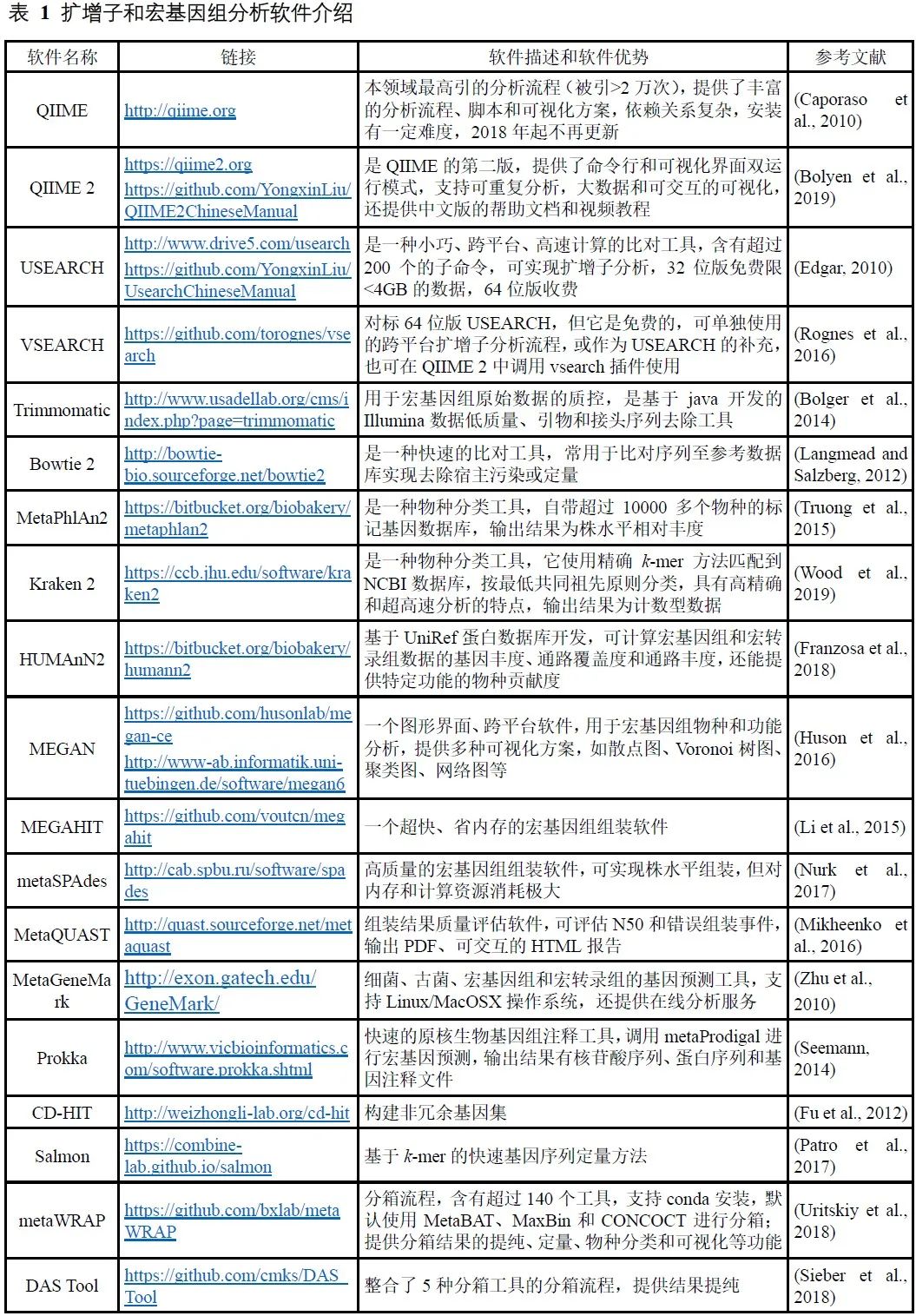
表 1 扩增子和宏基因组分析软件介绍

注:点击以上软件名链接为全文解读,描述中的链接可查看中文实战教程。
4.统计和可视化
扩增子和宏基因组分析流程最重要的输出文件是物种和功能组成表(图2/3)。研究人员可以使用这些分析技术回答的科学问题包括:微生物群中存在哪些微生物?不同组在α和β多样性上是否存在显著差异?哪些物种、基因或功能通路是各组的生物标记?为了回答这些问题,需要从整体和细节的角度熟悉统计分析和可视化方法。整体可视化可用于探索特征表中α/β-多样性和物种分类学组成的差异。细节分析可能涉及生物标记识别、相关性分析、网络分析和机器学习等(图3)。我们将在下文讨论这些方法,并提供示例和参考资料来帮助理解这些分析过程(图3和表2)。
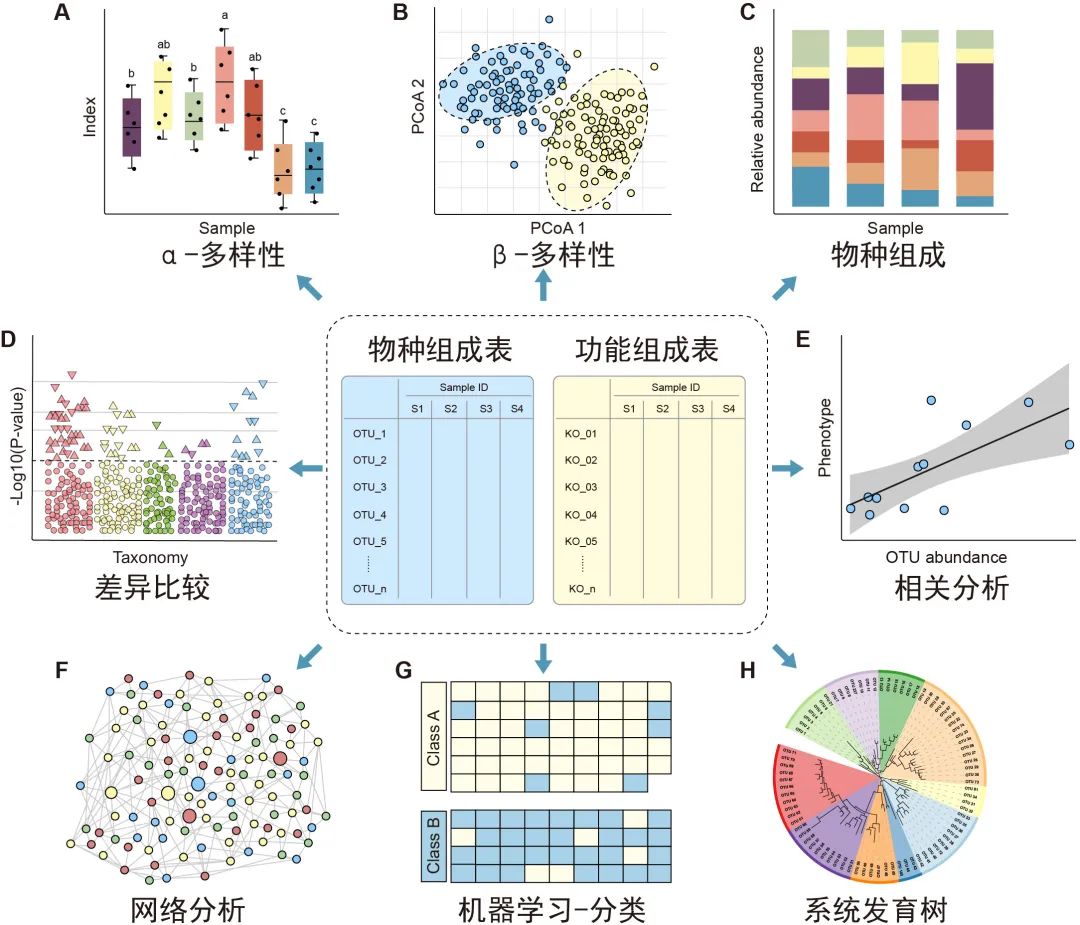
图3.特征表统计和可视化方法概览:微生物组特征表的下游分析包括 α 多样性(A)、β多样性(B)、物种组成(C)、差异比较(D)、相关分析(E)、网络分析(F)、机器学习分类(G)/回归以及使用树图(H)展示的系统发育树或分类层级树。有关更多详细信息请参见表2。
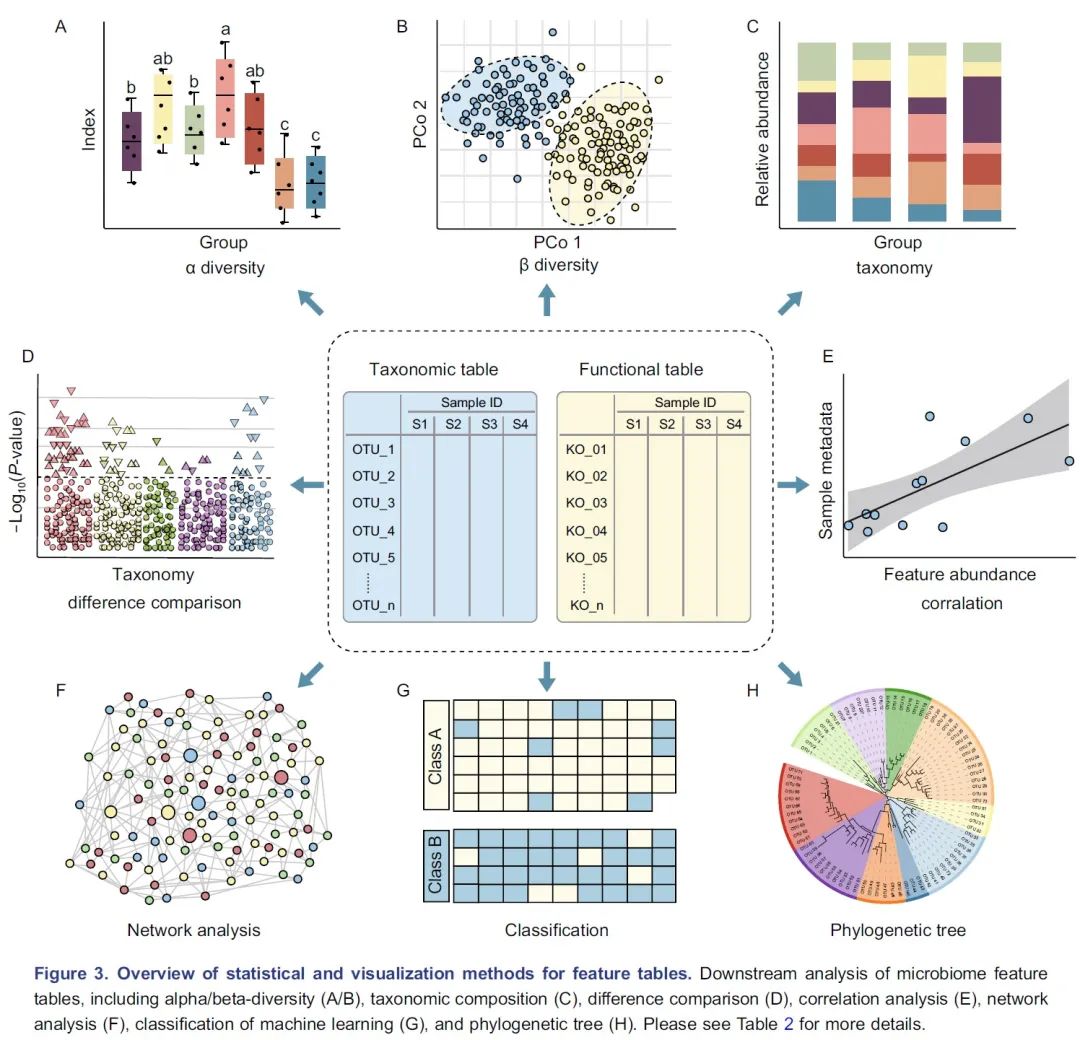
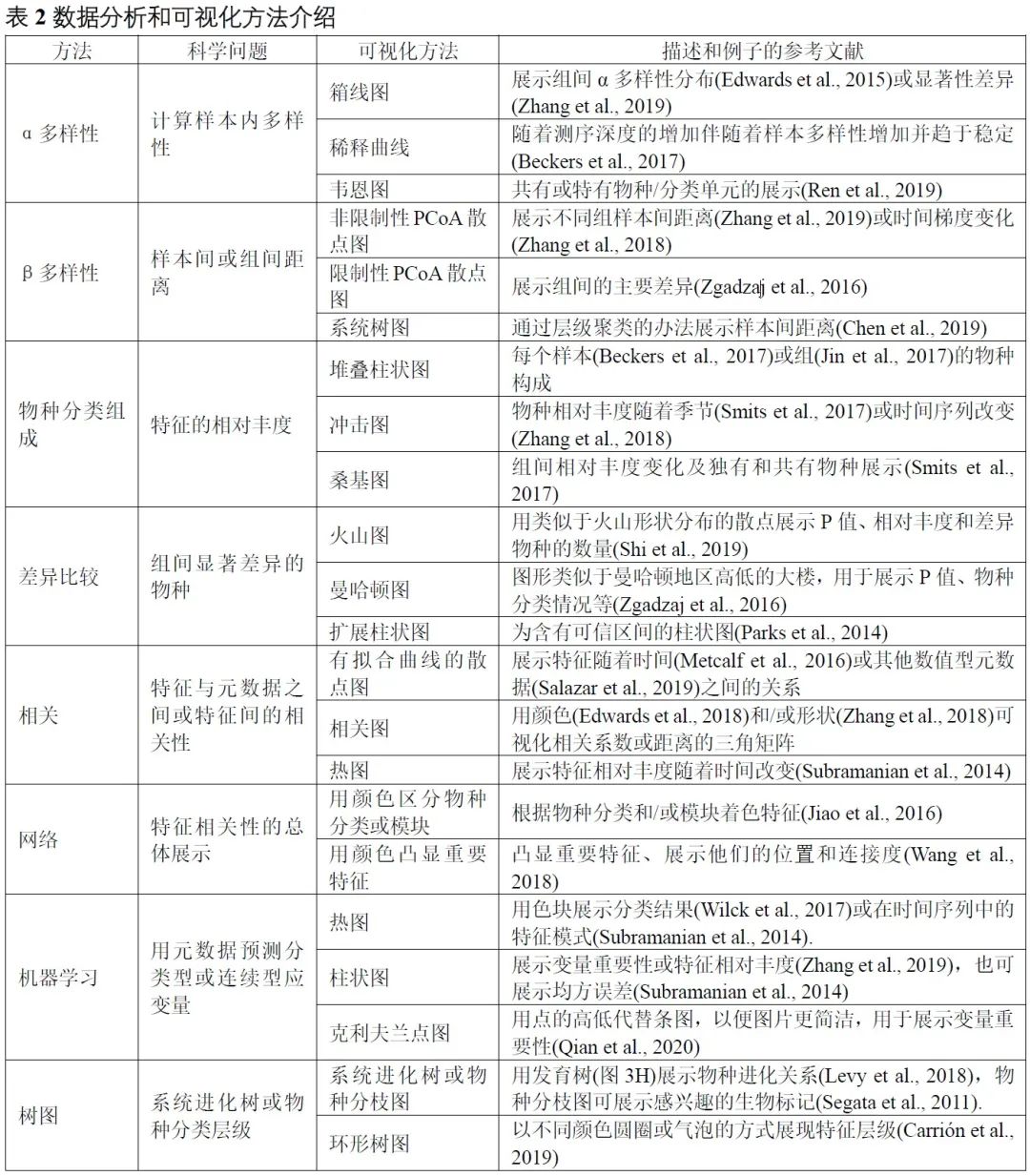
表2. 数据分析和可视化方法介绍
α多样性评估样本内的多样性,包括物种数量丰富度(richness)和均匀度(evenness)的测量。可以计算α多样性的软件包括QIIME、R语言的vegan包和USEARCH等。通常使用箱线图直观地比较每组样品的α多样性值(图3A)。可以使用方差分析(ANOVA)、Mann-Whitney U检验(2组比较的秩和检验)或Kruskal-Wallis检验(≥3组的秩和检验)来统计评估组间的α多样性差异。注意,如果将每个组进行两次以上的比较,则应校正P值。表2中介绍了其他有关α多样性指数的可视化方法。
β多样性评估样本之间微生物组的差异,通常将其与降维方法结合使用以便顺利实现可视化。降维方法有主坐标分析(PCoA)、非度量多维尺度(NMDS)或限制性主坐标分析(Constrained PCoA)等。这些分析可以在R包vegan中完成,并以散点图的方式可视化(图3B和表2)。这些β-多样性指数之间的差异可以用置换多元方差分析(PERMANOVA)进行计算,计算工具以vegan包中的adonis()函数最常用。
物种的分类学组成(taxonomic composition)描述了微生物群落中存在的微生物类群,通常使用堆叠柱状图进行可视化(图3C和表2)。为简单起见,微生物通常以门或属的水平在图中展示。
差异比较用于识别组间特征差异,这些特征可以是物种、基因或通路等。可以使用的检验方法有Welch’s t检验、Mann-Whitney U检验、或Kruskal-Wallis检验等,也可用ALDEx2、edgeR、STAMP或LEfSe等包或软件完成分析。差异比较的结果可以使用火山图、曼哈顿图(图3D)或扩展柱状图(表2)可视化。注意,由于组成型特征的相对丰度增加的同时伴有其他特征减少,这种数据的分析易于产生假阳性。研究者已经开发了几种方法来获得样品中的分类学绝对丰度,例如HTS结合流式细胞计数和“HTS+内参质粒+qPCR”的方法。
相关分析用于揭示特征和样本元数据(sample metadata)之间的关联(图3E),常用于识别分类群与环境因素(例如pH值、经度和纬度)或临床指标之间的关联,或用于识别在时间序列中影响微生物群和动态分类群的关键环境因素。
网络分析从整体角度探讨了特征的共现性关系(图3F)。相关网络的属性可能表示同时出现的分类单元或功能路径之间的潜在相互作用。相关系数和P值的计算可使用语言R中的cor.test()函数,或者用专门为稀疏型微生物组数据开发的SparCC包分析。网络的可视化和分析还可以使用R语言igraph包、CytoScape、Gephi等软件实现。网络分析可参考几个很好的例子,例如探索门或模块分布的研究(Fan et al., 2019)或显示时间序列上趋势的研究(Wang et al., 2019)。
机器学习是人工智能的一个分支,可以从数据中学习、识别模式并做出决策(图3G)。在微生物组研究中,机器学习用于特定特征的物种分类、β多样性分析、分箱和组成分析(compositional analysis)。常用的机器学习方法包括随机森林,Adaboost和深度学习等,他们可以用于“分类”和“回归”(表2)。注:“分类”是指应变量为分类变量,比如患病和未患病;“回归”是指应变量为连续型变量,如时间。
树图被广泛用于微生物组系统发育树(图3H)或物种层级分类注释的可视化。扩增子的代表性序列均为同源序列,非常适合于系统发育分析。我们建议使用适合大数据的IQ-TREE快速构建高可信度的系统树,并使用iTOL实现在线可视化。推荐使用R脚本table2itol(https://github.com/mgoeker/table2itol)快速生成iTOL格式的树注释文件 。此外,我们还推荐使用GraPhlAn构建高颜值的系统发育树或层级分类分支图(Cladogram)。
此外,研究人员可能对探明微生物来源感兴趣,以解决诸如肠道菌群起源、河流污染来源以及法医学检测等问题。FEAST和SourceTracker用于揭示微生物群落的起源。如果研究人员希望关注宿主遗传信息与微生物之间的调控关系,则全基因组关联分析(GWAS)可能是一个不错的选择。
5. 可重复分析
可重复分析要求研究人员在出版论文的同时提交他们的数据和代码,而不仅仅是描述他们的方法。可重复性对于微生物组分析至关重要,因为如果没有原始数据、详细的样本元数据和分析代码,就不可能重现结果。如果读者可以运行代码,他们将更好地理解研究中所做的事情。我们建议研究人员使用以下步骤共享其测序数据、元数据、分析代码和详细的统计报告。
5.1 将原始数据和元数据上传至数据中心
扩增子和宏基因组测序产生大量原始数据。通常,原始数据必须在论文发表期间上载到数据中心,例如NCBI、EBI或DDBJ。近年来在中国也建立了几个数据中心以提供数据存储和共享服务。例如,中国科学院北京基因组研究所建立的组学原始数据归档库(GSA)具有很多优势(表3)。我们建议研究人员将原始数据上传到表3中的数据中心之一,这不仅可以提供数据备份,还可以满足论文发表要求。诸如《Microbiome》等期刊都要求在投稿之前将原始数据存储在数据中心。
5.2 与其他研究人员共享数据分析代码
分析代码可以帮助审稿人或读者评估实验结果的可重复性。我们在 https://github.com/YongxinLiu/Liu2020ProteinCell 提供了用于扩增子和宏基因组分析的示例分析流程。此外,还应提供分析中使用的运行环境和软件版本,以确保可重复性。如果使用Conda来部署软件,则命令 “conda env export -n environment_name > environment_name.yaml” 可以生成一个文件,其中包含所用软件及版本信息。对于不熟悉命令行的用户,可以使用Qiita、MGnify和gcMeta等在线服务器进行分析。但是,在线服务器分析方式的灵活性差强人意,因为他们提供的可调节步骤和参数较少。
5.3 提供详细的统计和可视化报告
用于特征表统计分析和可视化的工具包括Excel、GraphPad和Sigma plot,但是他们是商业软件工具,而且较难快速重现结果。我们建议使用诸如R Markdown或Python Notebooks之类的工具来记录所有分析代码和参数,并将他们存储在诸如GitHub之类的版本控制管理系统中(表3)。这些工具是免费、开源、跨平台的,并且易于使用。我们建议研究人员在R markdown文件中记录微生物组的所有统计分析和可视化方法。RMarkdown文档可以包含代码、表格、图片,支持输出为PDF/网页/Word格式报告方便阅读。这种工作模式将大大提高微生物组分析的效率,并使分析过程透明且易于理解。R可视化代码可以参考R Graph Gallery(表3)。可以将分析用的输入文件(特征表+元数据)、分析代码(* .Rmd)和输出结果(图、表和HTML报告)上载到GitHub,这将方便同行重复您的分析或重用您的分析代码。ImageGP(http://www.ehbio.com/ImageGP )提供了20多种统计和可视化方法,对于没有R语言背景的研究人员而言是一个不错的选择。
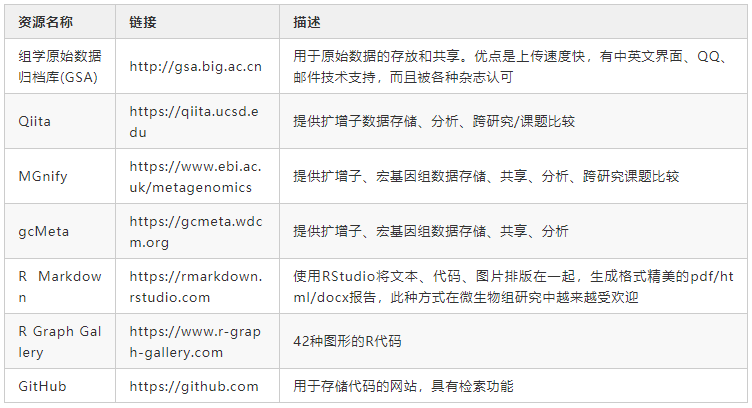
表3 提供可重复分析的网站和工具
6. 注意事项和展望
值得注意的是,实验操作对研究结果的影响远大于选用的分析方法。最好将详细的实验过程记录为元数据,例如采样方法、时间、位置、操作员、DNA提取试剂盒、批次、引物和标签序列等。元数据可以用于下游分析,而且可以帮助研究者确定是否这些操作差异导致了假阳性结果。实验中应收集的元数据等信息请参阅“标记基因序列的最少信息标准(MIMARKS)和宏基因组序列的最少信息标准”(Field et al., 2008; Yilmaz et al., 2011),“细菌和古菌单个扩增基因组(MISAG)和宏基因组组装的基因组(MIMAG)的最少信息标准”(Bowers et al., 2017),以及“未培养病毒基因组的最少信息标准”(Roux et al., 2019)。
一些特定的实验步骤可提供微生物组分析的独特视角。例如,开发和使用去除宿主DNA的方法可以有效增加植物内生菌和人类呼吸道感染样品中真实微生物组的比例。叠氮化丙锭可以物理去除土壤中大量残留的DNA。此外,当使用微生物含量较低的样品时,研究人员必须格外小心,以免由于污染而导致假阳性结果。对于这些情况,可使用不含DNA的水作为阴性对照。在人类微生物组研究中,个体之间的主要差异是由于饮食、生活方式和药物使用等因素造成的,遗传对于微生物组的影响小于2%。- Nature: 你的肠道菌群是遗传自你父母还是后天
随机(鸟枪)宏基因组测序可以深入了解菌株水平的微生物群落结构,但很难恢复高质量的全基因组。单细胞基因组测序显示了在微生物组研究中非常有前途的应用。基于流式细胞仪和单细胞测序,MetaSort可以从分选的亚宏基因组样品中恢复高质量的微生物基因组(Ji et al., 2017)。最近开发的第三代测序技术已用于宏基因组分析,例如太平洋生物科学(PacBio)的单分子实时(SMRT)测序和牛津纳米孔(ONT)测序平台。随着测序数据质量的提高和成本的降低,这些技术将引发微生物组测序领域的技术革命,并将微生物组研究带入一个新时代。
7. 结论
在这篇综述中,我们讨论了各阶段分析扩增子和宏基因组数据的方法,包括测序方法的选择、分析软件和流程、统计分析和可视化以及可重复分析的实施等。其他方法,如宏转录组、宏蛋白组学和宏代谢组学等可提供微生物组的动态信息,但是由于这些方法成本高以及需要复杂的实验和分析方法且不够成熟,尚不及扩增子和宏基因组如此广泛应用,所以未在本文的讨论范围之内。将来随着这些技术的进一步发展,对于微生物组研究的视角将更全面。
参考文献
Yong-Xin Liu, Yuan Qin, Tong Chen, Meiping Lu, Xubo Qian, Xiaoxuan Guo & Yang Bai. (2020). A practical guide to amplicon and metagenomic analysis of microbiome data. Protein & Cell 41, 1-16, doi: https://doi.org/10.1007/s13238-020-00724-8
原文链接:http://bailab.genetics.ac.cn/pdf/Liu-2020-ProteinCell.pdf







