编者按:
在过去的 15 年里,微生物组学领域取得了前所未有的进展,我们获得了许多关于微生物组的新知识和新见解,然而,想要把这些新知识和新见解融入到临床实践,我们依然面临着许多挑战和限制。
今天,我们特别编译发表于 Trends in Molecular Medicine 杂志上题为“Breakthroughs and Bottlenecks in Microbiome Research”的文章,希望本文能够为相关的产业人士和诸位读者带来一些启发和帮助。
与所有哺乳动物一样,人类也携带着多种共生微生物(被称为微生物组)。在过去的 15 年内,我们见证了微生物组学研究取得了前所未有的突破,然而,微生物组学研究仍面临着许多限制、瓶颈以及挑战。这些问题需要年轻一代的研究人员通过振奋人心的基础研究和生物医学研究来一一解决。

高通量测序技术为观察复杂的共生群落提供了无与伦比的手段。像人类微生物组计划(HMP)、MetaHIT(人体肠道宏基因组计划)、American Gut(美国人肠道菌群研究计划)、Flemish Gut(比利时人肠道菌群研究计划)这样的研究项目已经为我们揭示了微生物组与人类健康结果之间的联系1。
尽管二代测序技术(NGS)提供了很多机会,但它也暴露出基于测序的群落分析存在的局限性。一个急迫的问题是,定量微生物组学和宏转录组是否比单独研究微生物组组分更能揭示微生物组?是否需要更多的微生物组序列?
事实上,人类炎症性肠病(IBD)的开创性研究提供了证据,除了微生物比例组成的改变外,微生物载量/负荷可能是微生物组发生变化的关键驱动因素2。
某些微生物尽管在宏基因组层面丰度较高,占主导地位,但其几乎没有基因表达(图 1)3。与微生物相关的人类疾病中,疾病表型往往仅与微生物菌株的某种亚型相关。新兴的方法使基于高通量宏基因组测序进行菌株水平分析成为可能4。(详见 Box)
然而,宏基因组测序从根本上就存在局限性,因为它无法直接反映群落的功能活动。为了准确地为与微生物组结构相关的人类健康结果建立模型,我们急需将精力从进行不同数据类型的孤立研究转移到结合了不同数据类型的系统性研究,后者包含了大量协变量。
因此,我们需要额外的多组学数据来完整地描述微生物组。例如,将群落 RNA 丰度(宏转录组学)、蛋白质(蛋白组学)和代谢物(代谢组学)整合起来描述微生物组的全貌5。
对多种不同的数据类型(即二代测序技术和质谱技术)进行综合分析,这对于统计而言挑战很大,尤其是当其中每一类数据都是由不同分布的相对丰度数据组成的。因此,想要将此类复杂而微妙的科学结果直接转换为临床实践,应保持谨慎。
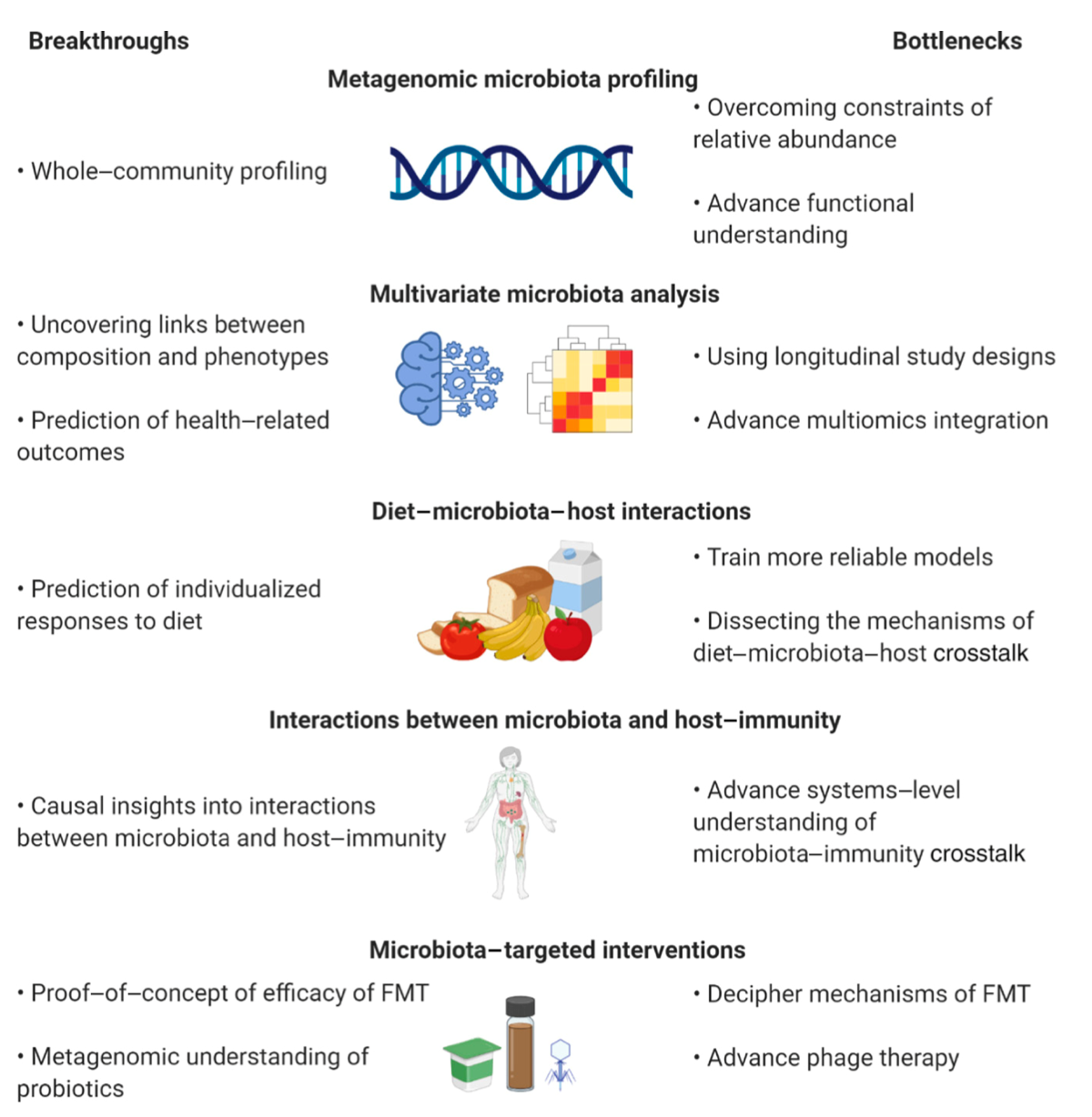
图 1 微生物组研究的突破和瓶颈 该图描绘了微生物组研究在不同领域中的重大突破(左)和瓶颈(右)。FMT,粪菌移植。
个体间的高度差异性:一个典型的肠道宏基因组学样本包括了从几百个到上千个不同的分类单元,不同的个体在这些分类单元的数量上表现出高度的可变性。种族、地理位置以及生活方式上的差异只是造成差异的一些主要影响因素,这些差异限制了微生物组结论的普遍性。理想的统计分析需要较大的样本量,来克服这种可变性并得出有意义的结论。
依赖于相对丰度:不依赖于培养的方法(如二代测序技术)存在一个重大局限性,即不能提供可靠的绝对丰度或者功能的信息。基于培养的方法可以提供有关微生物绝对丰度的信息,但由于目前的技术限制(需培养的微生物数量以及一些不可培养微生物),其表征复杂微生物群落(如哺乳动物的肠道微生物)的能力有限。
依赖于粪便样品:收集粪便是一个相对来说方便且无创的方法。然而,粪便微生物组的组成或功能与胃肠道中的微生物组却不尽相同。
着眼于细菌:在微生物组学研究当中经常被忽视的其他共生微生物(如真菌、病毒),越来越被认为与健康和疾病相关。
着眼于肠道:对皮肤、泌尿/生殖道、唾液或肿瘤等生物量较低的样本进行微生物组分析,在技术上具有挑战性,因为它们容易因标本采集和处理过程中的污染而产生假阳性结果。因此,此类样本较少纳入微生物组研究。

在影响肠道微生物组结构的因素中,饮食至关重要。饮食习惯是造成人与人之间肠道微生物组不同的重要原因。反之,肠道微生物组也起着‘信号中枢’的作用,向宿主产生丰富的与饮食相关的信号。
在过去几年内,可能不存在‘一刀切’的饮食建议以使人类获得健康益处的观点变得越来越重要。
一些研究表明,利用基于微生物组的机器学习模型预测个体对某些食物的特定反应是可行的6,7。机器学习和其他人工智能方法需要利用微生物和临床特征数据集来训练模型,以学习特定食物对给定的健康相关结果的影响。
原则上,任何一类可量化数据都可用于实现这一目的,而不必了解这一预测背后复杂的机理联系。
将个性化饮食的概念带入现实世界应用的关键挑战是建立足够大的队列来收集足够多的数据,使得微生物组和其他数据能够进行足够深入和全面的剖析,以训练可靠的模型。
同时,还存在另一个同样重要的步骤——对预测特定生理反应的特征进行机制剖析。这对于制定预防或治疗干预措施具有很高的价值,正如对患有肠病的营养不良儿童的多组学调查所显示的那样8。
关于该研究《热心肠日报》做过相关报道:
NEJM:小肠疾病相关的儿童发育不良,可能是小肠菌群有问题
New England Journal of Medicine[IF:74.699]
① 分析36名环境肠功能障碍(EED)患儿的十二指肠微生物群,鉴定出14个不属于典型肠道病原体的EED核心细菌类群;② 这些细菌(某韦荣球菌、某链球菌和Rothia mucilaginosa等)的绝对水平,与患儿生长负相关,与参与免疫炎症应答的十二指肠蛋白(如LCN2)正相关,且在患儿粪便中的含量不同于健康儿童;③ 和EED核心菌相关的十二指肠蛋白,与血浆REG3A等显著相关;④ 定植EED十二指肠菌群的部分分离菌(含大部分EED核心菌)可引起小鼠小肠病变。
Duodenal Microbiota in Stunted Undernourished Children with Enteropathy
2020-07-23, doi: 10.1056/NEJMoa1916004
【主编评语】环境肠功能障碍(EED)是一种小肠疾病,被认为是儿童发育不良的潜在原因,但由于小肠样本难以获取,人们对这种疾病还所知有限。New England Journal of Medicine杂志最新发表了Jeffrey Gordon团队主导的一项研究,对孟加拉80名经活检确诊为EED的营养干预无效的发育不良儿童的血浆和十二指肠蛋白组、十二指肠组织病理,以及其中36名患儿的十二指肠微生物组等进行分析,鉴定出与发育不良相关的14个EED核心十二指肠细菌类群,以及潜在的十二指肠和血浆蛋白标志物。结合悉生小鼠模型,该研究表明这些EED菌群可能通过促进十二指肠炎症应答,参与EED的发生发展,从而导致发育不良。这些发现为调控小肠菌群来治疗EED相关儿童发育不良的干预策略,提供了支持证据。(@mildbreeze)

个体共生微生物种类作为临床诊断手段也有很大的前景。对粪便或血液进行宏基因组测序在感染或者恶性肿瘤方面具有强大且无创的诊断能力。例如,在感染的情况下,与作为金标准的培养方法相比,对体液进行宏基因组测序既能加快病原体的检测速度,还可能具备更高的诊断灵敏度9。
及时的病原体检测可以缩小抗菌药物的使用范围,减少经验性抗生素的广泛使用,最大限度地减少抗生素耐药性的出现——一个全球性的公共卫生威胁。
目前,在一些医疗中心,二代测序技术平台还没有被广泛使用,要想将其应用到临床护理中,需要在样本采集和数据分析方面进行一些方案调整,使其在临床应用中更易于医护人员的理解并且要具有一个患者可负担的价格。
微生物组研究大多集中于细菌群落成员,主要是由于研究其他种类的共生微生物面临着巨大的技术挑战。然而,新出现的证据表明,原生动物、真菌、古菌和病毒在稳态和疾病中发挥着关键作用。
噬菌体是一种采用宿主特异性方式来攻击细菌的病毒,自发现噬菌体以来,已经过去了 100 多年。然而,在宏基因组水平上研究噬菌体的可重复方案在最近才出现。
多重耐药细菌感染所带来的威胁迫在眉睫,这使人们重新燃起了对噬菌体作为传统抗生素替代品的兴趣。
噬菌体和肠道内细菌之间的种群动态变化规律是最难以捉摸的,也是一个正在研究的领域10。推进针对感染性疾病及生活方式相关疾病的噬菌体疗法的基本挑战在于识别和分离对宿主具有有效性并且基因组安全的关键噬菌体联合体。
宏基因组的迅猛发展重新激起了人们对于粪菌移植(FMT)这一‘新的传统工具’的兴趣。超过 350 项已完成或计划进行的临床试验证明了人们对于粪菌移植的兴趣(美国国立卫生研究院 NIH,2020 年 12 月)。
迄今为止,关于 FMT 最可信的数据是用于治疗复发性艰难梭菌感染,另一个潜在的适应症是溃疡性结肠炎。目前,它的适用性只限于选定的常规治疗失败的患者11。
到目前为止,所有的随机对照实验都存在明显的局限性。因此,我们当前仍不能充分评估粪菌移植的有效性和安全性。一个不确定因素是供体将感染性病原体传给受体的可能性。另一个不确定因素是不同研究之间在患者选择和技术程序上存在异质性。
上述因素对解释粪菌移植的作用机制至关重要。一系列研究指出,除粪菌移植外,其他因素也可能是决定性的(如胆汁酸或噬菌体)12,13。这些因素的确定,将使我们突破相对简单的粪菌移植流程,进行更可控、更有力的精准干预。
宏基因组测序技术的突破让人们对益生菌——一种食用后可能会给健康带来好处的活的微生物——有了新认识14。
大量的研究剖析了各种市售益生菌菌种相关的临床结果。然而,表征不足,缺乏对潜在作用模式的机制理解,以及缺乏对预测反应的宿主因素的识别可能导致了参差不齐的试验结果,以及未被监管部门批准作为医疗干预手段。
当务之急是超越基于经验论来使用益生菌,并确定具有合理定义的有益菌群、促进其生长的饮食因素,以及精确定位具有有益效果的微生物衍生代谢物。新兴的技术将允许大规模挖掘这些与宿主生理学具有显著互作的代谢物15。
迄今为止,对人类微生物组的研究已经产生了许多有价值的观点与见解,但也产生了大量缺乏因果证据的相关性和观察性结果。虽然微生物组学研究在过去的几年里已经明显成熟,不再被认为是处于起步阶段,但它还没有实现彻底改变临床实践和预防菌群失调相关疾病的目标。
我们坚信,更加注重机制的微生物组学研究,更加致力于证明和解释因果关系,而非产生关联、相关性和预测的研究,将帮助微生物组学数据应用于精准医疗。
参考文献:
(滑动下文查看)
1.G., F. et al. (2016) Population-level analysis of gut microbiome variation. Science 352, 560–564
2.Vandeputte, D. et al. (2017) Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511
3.Schirmer, M. et al. (2018) Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 3, 337–346
4.Truong, D.T. et al. (2017) Microbial strain-level population structure & genetic diversity from metagenomes. Genome Res. 27, 626–638
5.Franzosa, E.A. et al. (2015) Sequencing and beyond: integrating molecular “omics” for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372
6.Zeevi, D. et al. (2015) Personalized nutrition by prediction of glycemic responses. Cell 163, 1079–1095
7.Berry, S.E. et al. (2020) Human postprandial responses to food and potential for precision nutrition. Nat. Med. 26, 964–973
8.Chen, R.Y. et al. (2020) Duodenal microbiota in stunted undernourished children with enteropathy. N. Engl. J. Med. 383, 321–333
9.Gu, W. et al. (2021) Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat. Med. 27, 115–124
10. Rasmussen, T.S. et al. (2020) Bacteriophagemediated manipulation of the gut microbiomepromises and presents limitations. FEMS Microbiol. Rev. 44, 507–521
11.Mullish, B.H. et al. (2018) The use of faecal microbiota transplant as treatment for recurrent or refractory Clostridium difficile infection and other potential indications: Joint British Society of Gastroenterology (BSG) and Healthcare Infection Society (HIS) guidelines. Gut 67, 1920–1941
12.Ott, S.J. et al. (2017) Efficacy of sterile fecal filtrate transfer for treating patients with Clostridium difficile infection. Gastroenterology 152, 799–811.e7
13.Zuo, T. et al. (2018) Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut 67, 634–643
14.Zmora, N. et al. (2018) Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell 174, 1388–1405.e21
15.Chen, H. et al. (2019) A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell 177, 1217–1231.e18
原文链接:https://www.cell.com/trends/molecular-medicine/fulltext/S1471-4914(21)00015-0?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1471491421000150%3Fshowall%3Dtrue
作者|Timur Liwinski, Avner Leshem, Eran Elinav
编译|Johnson
审核|617
编辑 | 笑咲






