


各位好,我是北京大学朱怀球。
我今天从生物信息学进行微生物组学分析的角度来谈谈肠道菌群的复杂性,所以我今天的报告题目叫《细微之中有“大道”》。所谓“大道”,就是我想跟各位介绍的“肠道菌群的复杂性”。
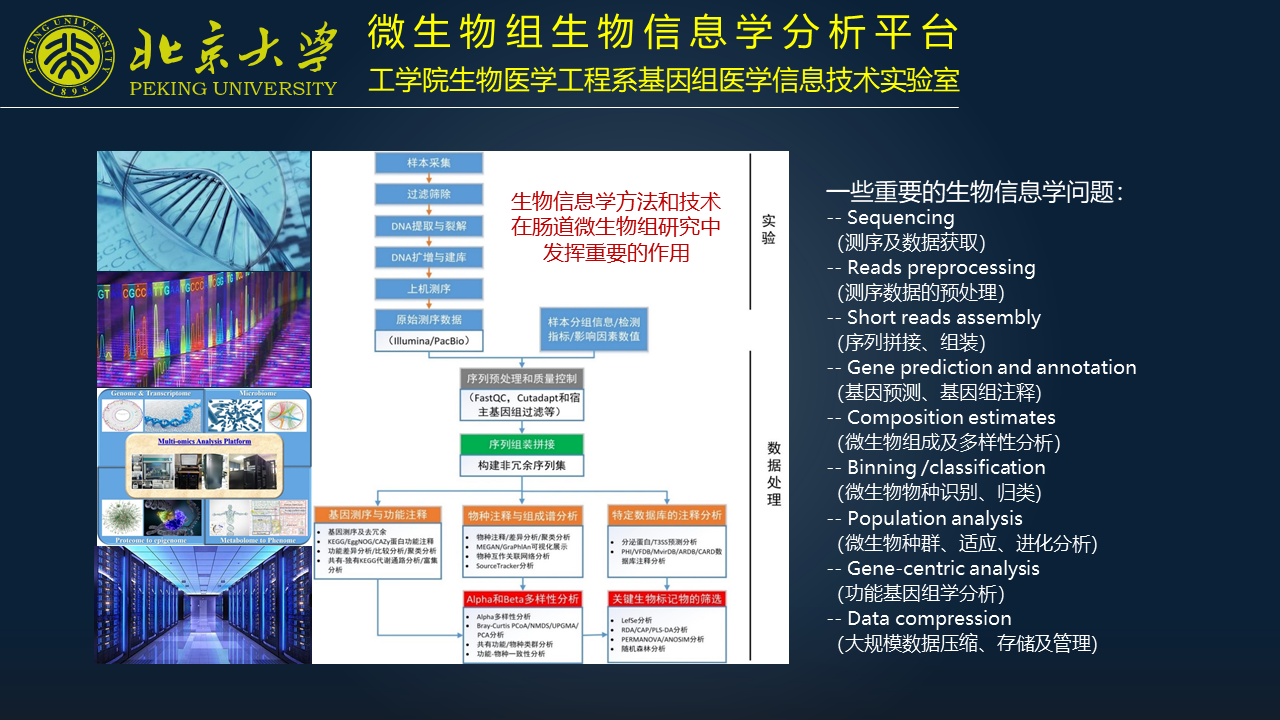
我的研究方向是生物信息学,我所在的实验室关注微生物组学当中的一些生物信息学的问题。实际上现在生物信息学的技术、方法在肠道微生物组研究中发挥着越来越重要的作用,涉及到很多重要的生物信息学的问题。
比如测序、数据获取、预处理,序列的拼接、组装,基因的预测、基因组注释,微生物组的组成和多样性分析,微生物物种的识别、归类,微生物种群的适应、进化分析,以及基于功能的分析和基于数据本身存储管理的问题等。
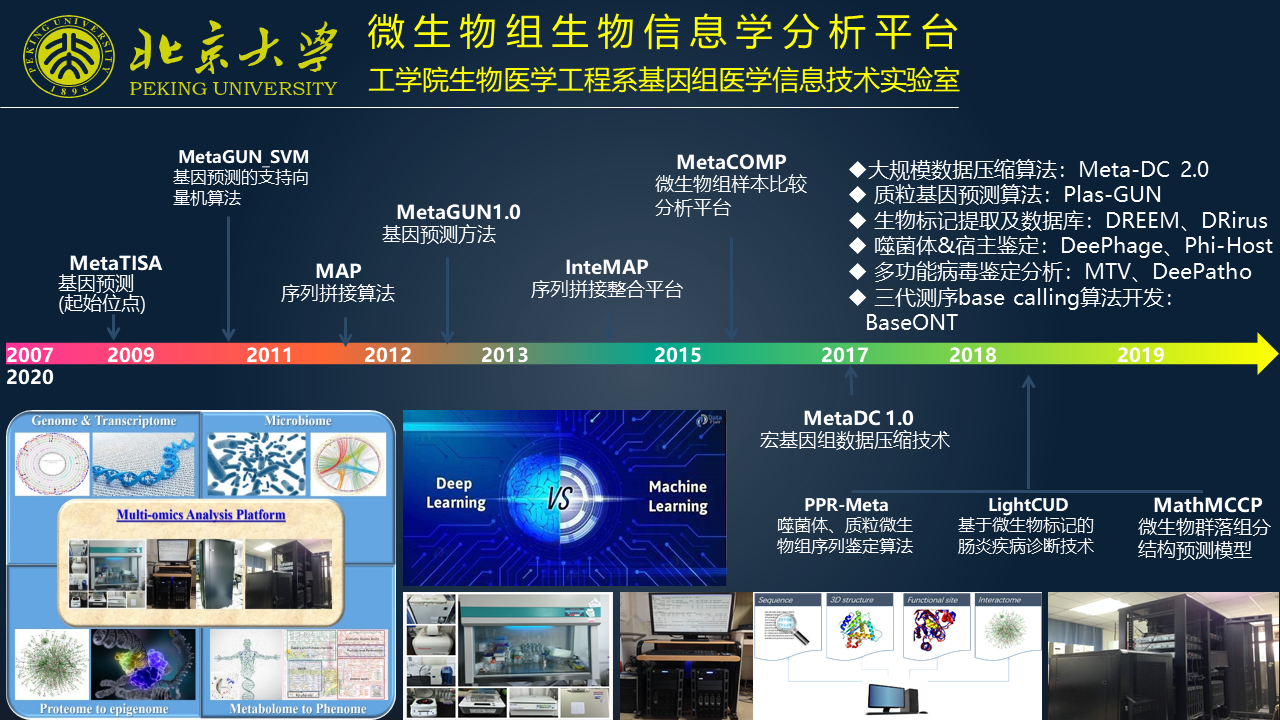
在这 10 多年以来,我们从最早的基因位点的识别到基因预测,到序列的拼接,种群结构的预测、分析等方面,也发展了一系列自主的方法和工具。

我想从微生物的群体智慧谈起。单个的微生物本身是微米量级的,但是当它们聚集在一起的时候,我们可以看到很多非常美的效应。比如说生物膜。它们的细胞相对位置是固定的,当生物膜在扩张的时候,会产生复杂的、有规律的结构,实现它的一个最优化(右图)。
作为更复杂的、群居的菌群,这种有规模的、宏观上的行为和结构越来越引起我们的关注。这也是我们常说的复杂系统的一个涌现性的性质。
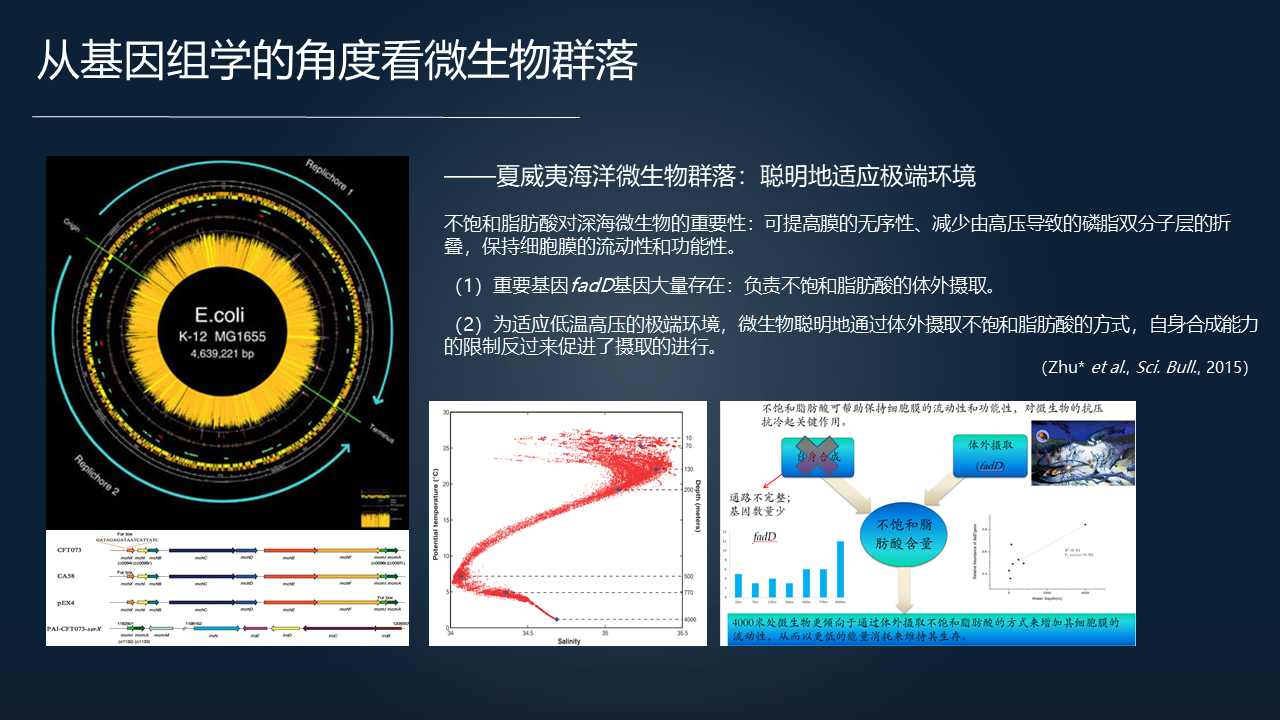
那么我们先看看微生物群落的一些非常有趣的特征。我们用夏威夷不同深度海洋微生物的全基因组测序去分析,在这里边可以看到非常有趣的现象。比如说在深渊区四千米深的高压、低温的环境下这种极端环境下,微生物群落很聪明的采取了一种适应这种极端环境的方式。
分析它的全基因组的时候,我们看到它合成短链脂肪酸、不饱和脂肪酸的代谢途径是缺失的。也就是它本身并没有合成不饱和脂肪酸的能力。但是不饱和脂肪酸对深海的微生物相当的重要,对保持细胞膜的流动、功能十分关键,使得它能够耐低温、抗高压。
同时我们又发现了它们大量的表达一种叫 fad 类型的基因。这种基因负责从体外摄取不饱和脂肪酸。而深海环境存在大量的生物遗骸资源,可以供微生物从体外摄取。所以这种的依赖外部获取脂肪酸的方式,体现了群落的 smart(聪明)的一个适应方式。
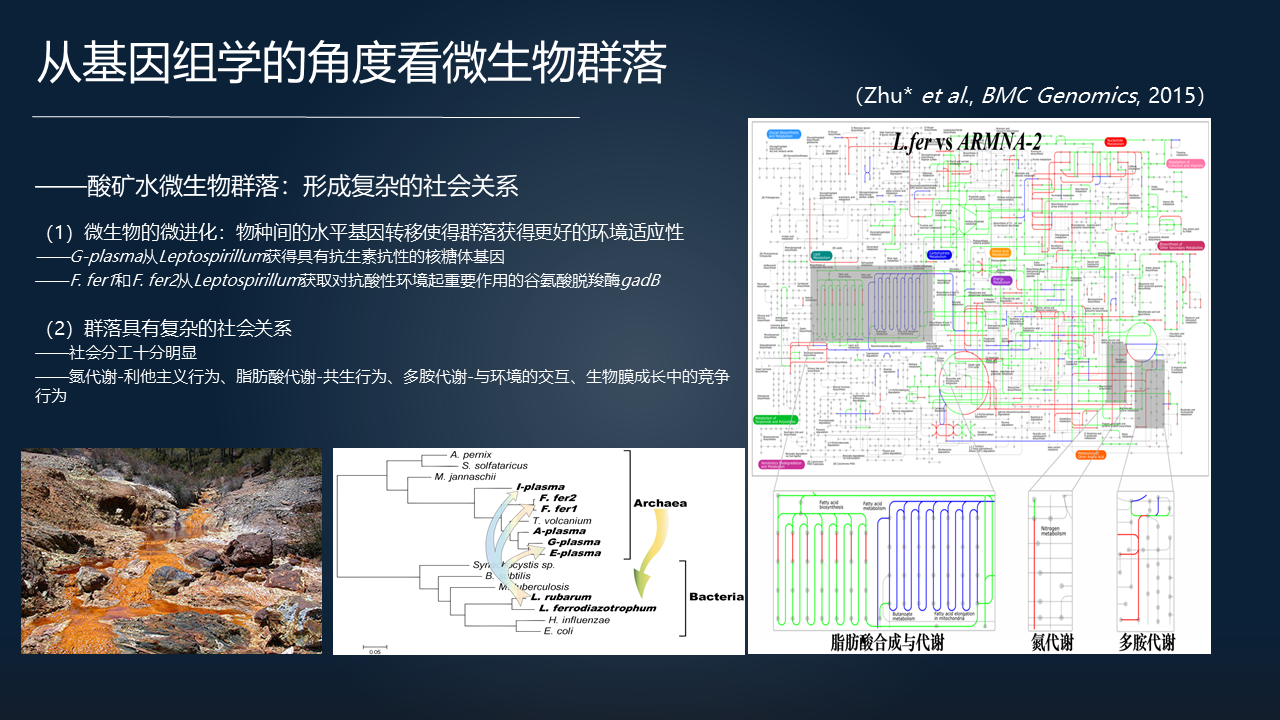
还有进一步的,我们也看到另一个很有意思的例子,那就是酸矿水的微生物群落。酸矿水应该是自然界中最强的一种酸性的环境,在这里边也生活着大量的微生物群落。通过对它们的分析也可以看到,在适应环境的时候,它们进一步的体现出一个复杂的社会关系。
这么一个性质,具体表现在它们可以通过基因水平的微进化、物种间通过水平基因转移来更好的适应环境。
比如说有些古菌通过向另外的细菌获取具有抗生素抗性的基因,来帮助它在群落里边成为数量占优的种群;有些古菌从其它的细菌里面去获取对抗酸性环境的基因,使得它能够更好的适应极端的环境。
全基因组的分析也让我们看到这种群落的社会分工十分明显。比如说,对于氮代谢的这种利他主义行为,对于脂肪酸代谢的共生行为,还有生物膜成长过程中竞争的行为等。
从环境微生物的角度去看,微生物虽然微小,但是作为群落,它们有所谓的这种群体智慧。
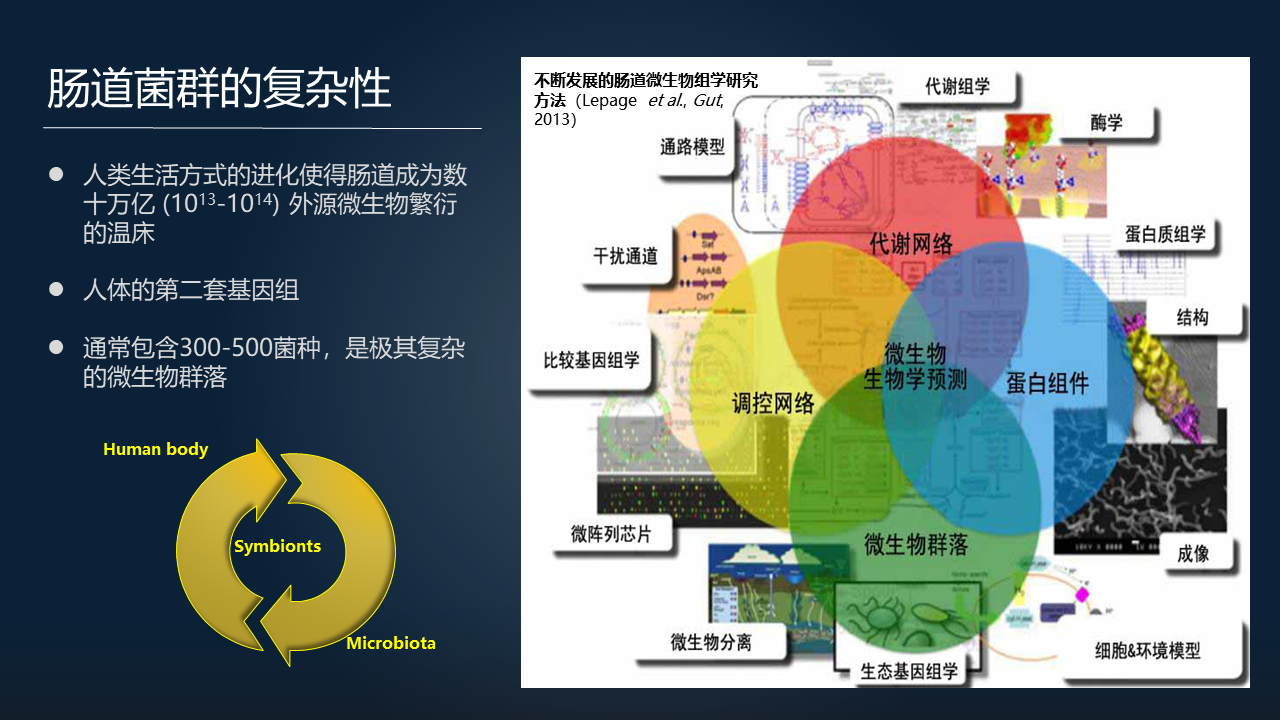
回到我们的肠道菌群,来看看肠道菌群的复杂性。首先我们都知道肠道菌群是我们人体的第 2 套遗传信息,它通常包含三百到五百种细菌,是非常复杂的微生物群落。
肠道微生物组学的研究方法从基因组学、转录组学,到蛋白质组学、代谢组学,从调控网络到代谢网络、结构等,研究方法在不断发展。
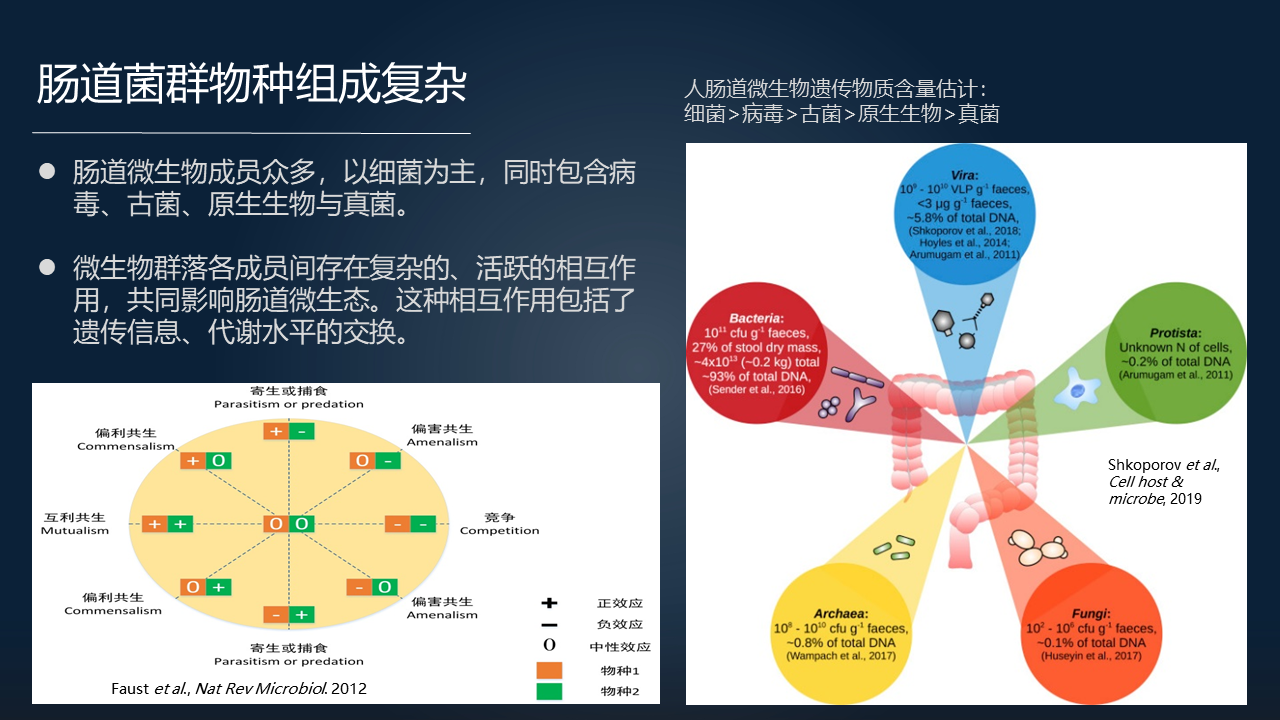
肠道菌群的复杂性首先是表现在物种组成上的,它的成员众多。很长时间以来人们主要关注其中的细菌,但肠道菌群也包含大量的病毒、古菌、原生生物、真菌等其它的遗传物质,而且这些成员之间又存在复杂的、活跃的相互作用,从而共同影响了肠道的微生态。
肠道菌群之间的相互作用包括遗传信息的交换、代谢、物质的交换等。对于肠道菌群来讲“什么是健康状态的肠道菌群,什么是偏离健康的,或者说处于紊乱状态的肠道菌群”是一个非常关键的问题。

怎么去定义它是否健康?我们希望从生物信息学入手,在理论上去回答这个问题。
我认为构造肠道微生物群落的生态有 5 个要素。
第 1 个要素是 Who(谁),也就说哪些细菌、噬菌体或者质粒、病毒是这个群落的成员。
第 2 个要素是 What(做什么),它们在群落里面的功能是什么?
第 3 个要素是 How to do(怎么做),通常表现在上面这些 Who 和 What 之间的相互作用。
第 4 个要素是群落作为一个整体(Whole),它们怎么样去适应或者改变外部的环境,包括宿主。
第 5 个要素是它是怎样随着环境的变化去演化的?比如说群落内部成员的快速的进化,以及群落作为一个整体,它适应环境的相对稳定的一个状态是否是这样那样的改变。
把这 5 个要素综合在一起去回答的话,我们就应该去构造一个所谓的泛微生物组(Pan-microbiome)的概念。Pan-Microbiome 的一个定义是从功能的水平,特别是从功能基因的水平来定义泛微生物组。与从分类的水平,属、种的定义不同。
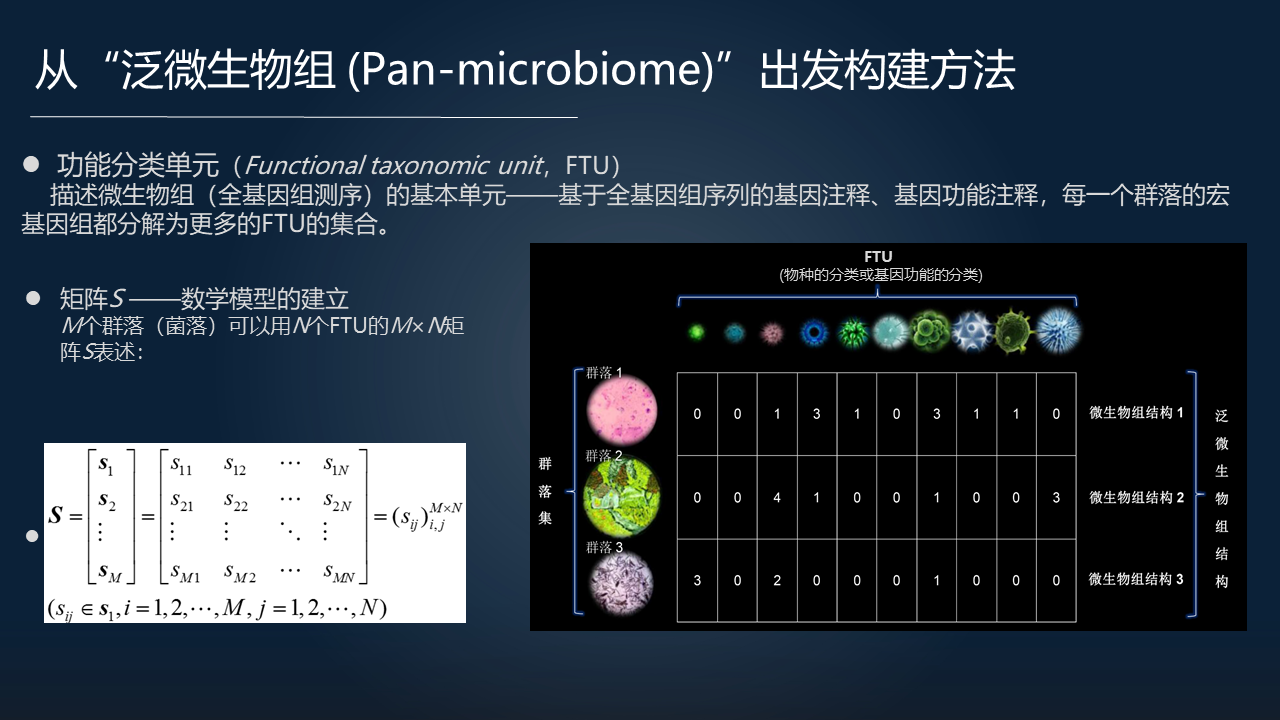

要解决这个问题,就需要我们先定义功能分类单元(FTU)。基于全基因组序列的基因注释把每一个群落所有的物种的基因组都分解为功能分类单元的集合,进而由此发展出数学方法。
我们首先需要构造一个由很多 FTU 构成的一个大型矩阵,然后在这个基础上进一步的去构建泛微生物组的数学模型。
笼统来讲,Pan-microbiome 是一系列具有共同性质的微生物组的总和。对 Pan-microbiome 的理解,包含对微生物功能特征的一个认识,也应该要包含对相互作用关系网络的认识。
这样基于 FTU 的概念,我们就可以通过严格的数学理论来定义 Pan-microbiome 的结构矩阵,以及在空间上去发展算法。所以实际上我们是把 Pan-microbiome 作为 Microbiome 概念的推广。
基于 FTU 的这个概念可以克服传统的统计学方法,必须得从单一样本结构去研究样本组的组间差异的局限性,而直接可以通过矩阵数学空间的相关性去解读样本的数据结构,并且去刻画样本组之间的差异。
在这个指导思想下,我们发展了一系列的算法,并在这个基础上进一步的去分析肠道菌群全基因组的数据。
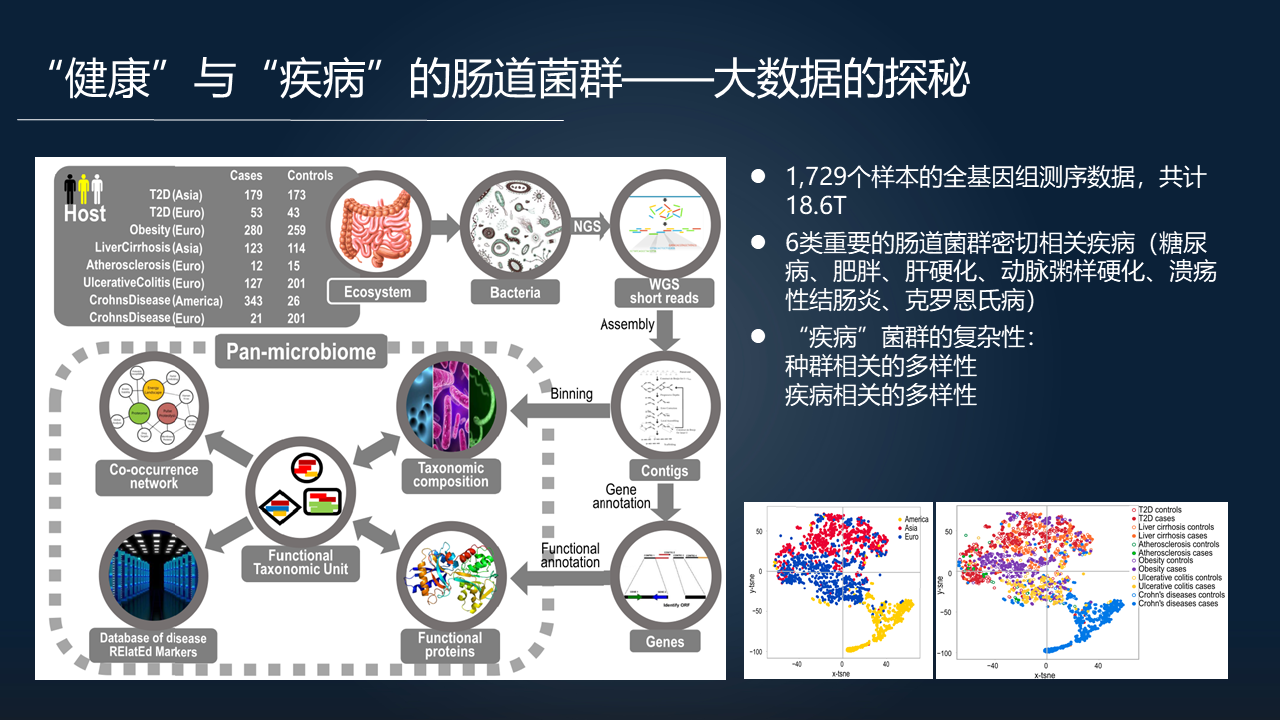
第一个有意思的例子就是我们用当时所能获取的六类重要的肠道菌群密切相关疾病的数据进行的研究。数据包括全基因组的测序数据,疾病则包括糖尿病、肥胖、肝硬化、动脉粥样硬化、结肠炎、克罗恩病等,一共包含了 1729 个样本,18.6 个 T。
通过大型计算去分析 1700 多个样本的结果是很有趣的,我们发现疾病相关的肠道菌群呈现出一系列的复杂性,首先表现在它跟它的宿主(人)的种群是相关的。美国人、欧洲人和亚洲人的肠道菌群差异比较明显,肠道菌群差异总体上跟种群是相关的。

其次,当我们看肠道菌群跟具体的疾病是否相关的时候(右下图),某一类疾病相关的肠道菌群是聚集在一起的。
这两种多样性交织,使得肠道菌群本身变得异常复杂。不同的人不同的疾病,同样的种群不同的病人,甚至相同的病人不同的时间段,菌群都体现出差异,体现出巨大的复杂性。
但与此同时,我们看到相对疾病相关的肠道菌群,它的对照组(健康的肠道菌群)共性则更为显著,就没有体现出那么强的种群的差异性、疾病的差异性和多样性。
因此我们把这种看到的健康的肠道菌群有更多的共性性质,概括成“幸福都是相似的,而不幸有多种多样的原因”。
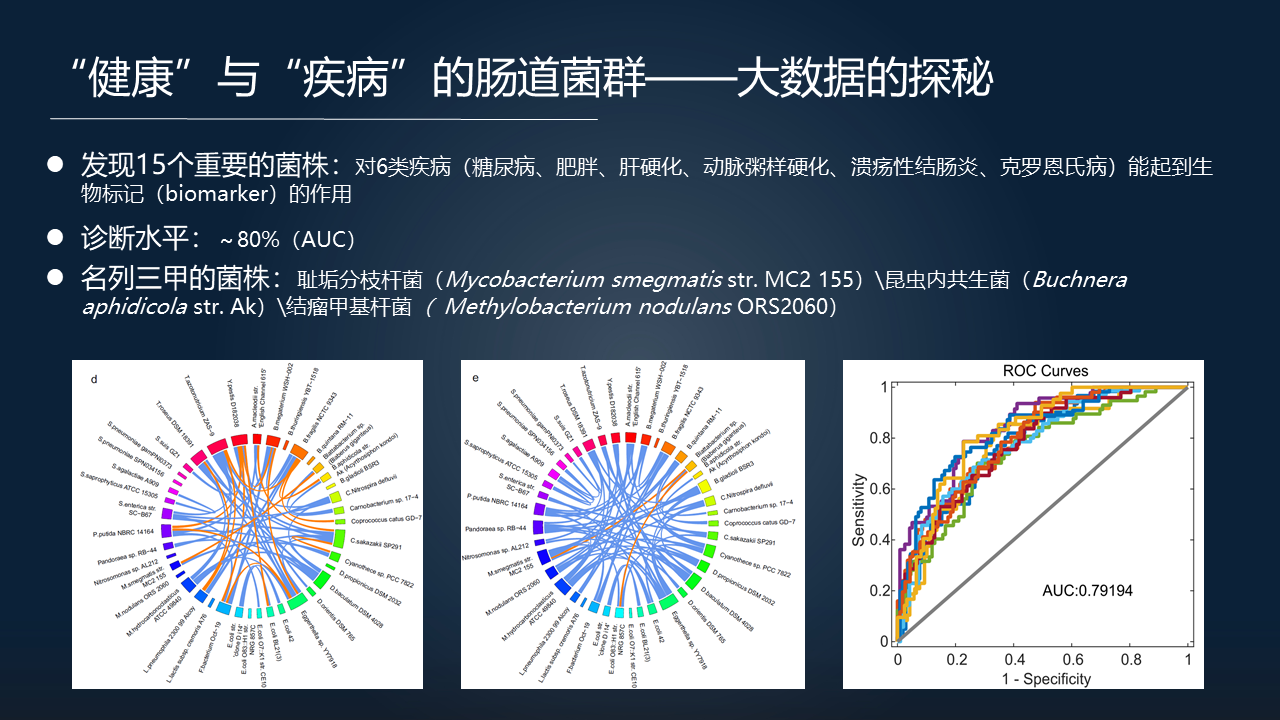
我们对这 6 种疾病进一步的进行数据挖掘,最终发现了 15 个非常重要的菌株。
这 15 个菌株能够起到一个 biomarker(生物标记)的作用。如果用这些 biomarkers 去做判别分析,诊断的 AUC 水平能够达到 80% 左右。这 15 个里面名列前三甲的菌株,包括大家能关注到的耻垢分枝杆菌、昆虫内共生菌、结瘤甲基杆菌等。
这些所挖掘出来的菌株可能都非常值得我们去进一步的关注。
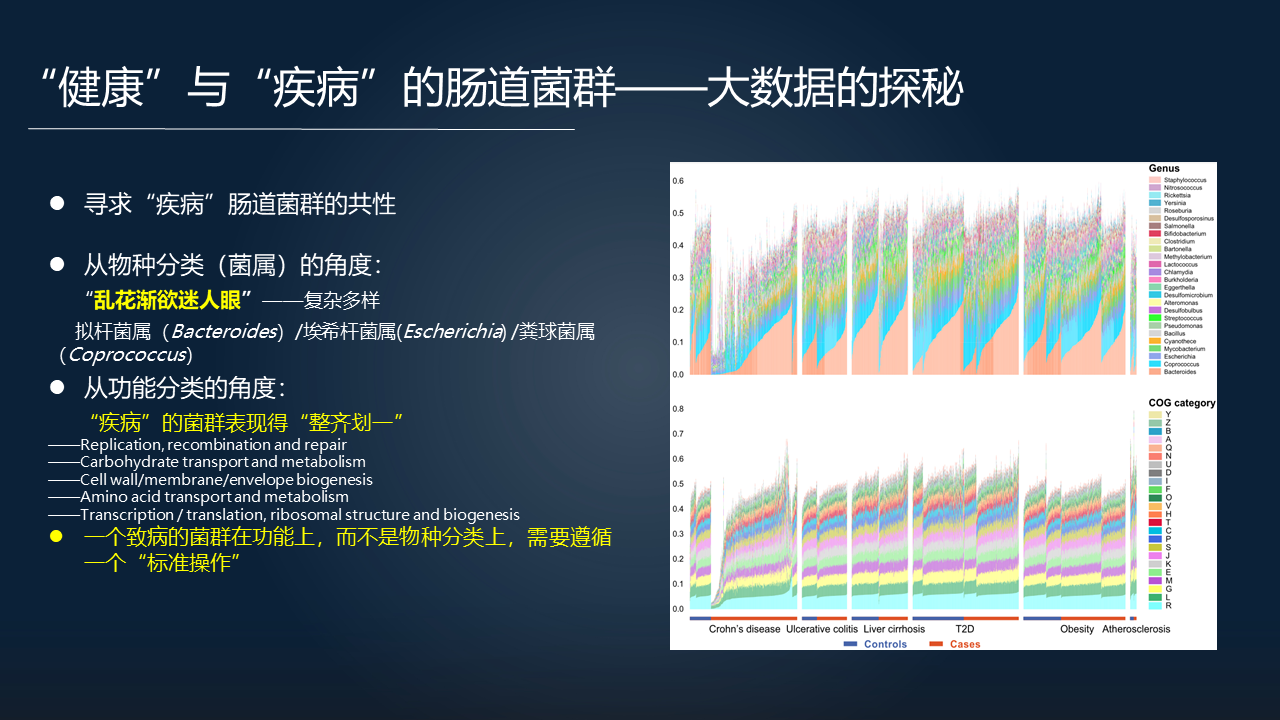
进一步的我们在想:疾病肠道菌群的共性是什么?我们去把疾病相关的菌群再做分析,如果从物种分类的角度去看的话(右上图),横坐标是六类疾病,疾病组和对照组之间体现出一种眼花缭乱的一个多样性,是复杂的。
当然我们可以看到里边拟杆菌、埃希杆菌、粪球菌在这六类疾病中都是差异显著的。但是把它们放在一起看,似乎还是多样性更加明显,差异性还是很大的。
相反,如果我们根据 FTU 从功能分类的角度去看的话(右下图),根据 COG 的功能分类的水平,把 FTU 计算得到的疾病组的菌群,相比上面的图来说,更加整齐划一。
我们可以从里面看到这些重要的差异显著基因的功能,包括复制、细胞膜等这些功能。因此我们是否可以得到一个结论,一个致病的菌群在功能上需要遵循一个标准的操作,而不是在物种分类上。
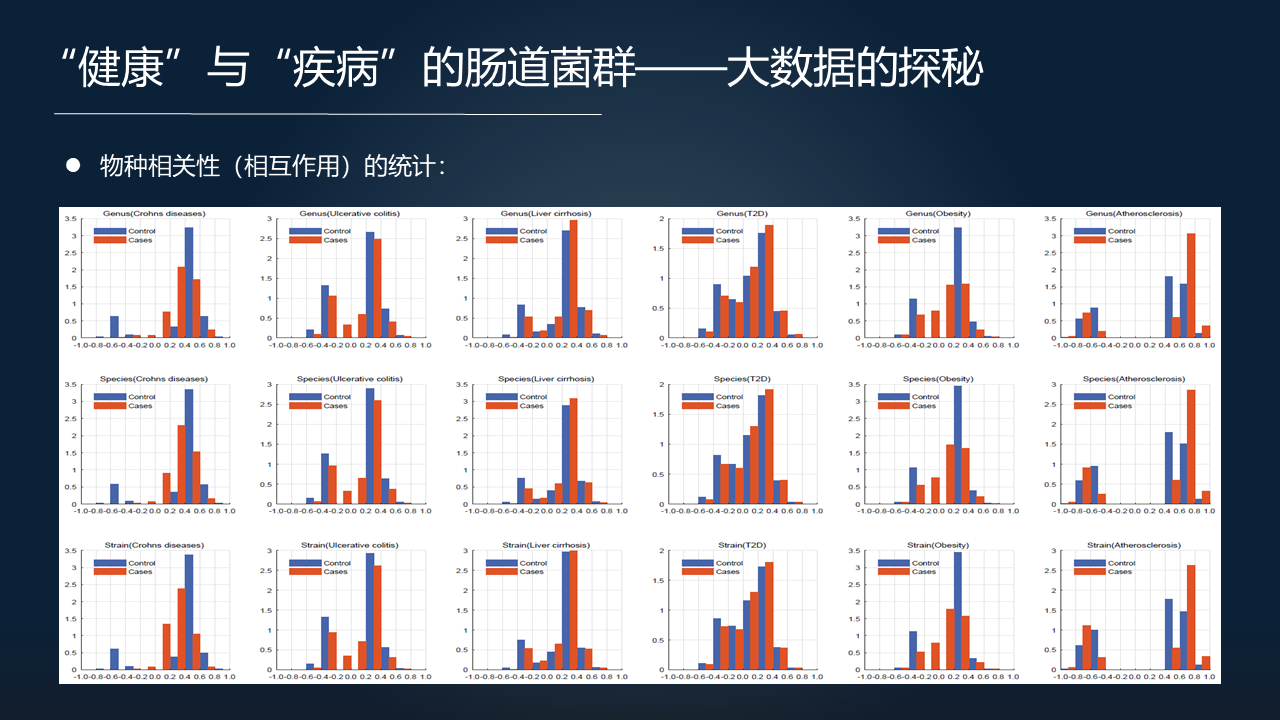
我们把 1700 多个样本按照疾病分别在不同水平上算完 FTU 以后进行比较,可以看到一个大致的趋势,也就是疾病中物种之间的相关性的数据。物种之间的相关性在生物信息学里面通常会用来代表物种之间可能存在的相互作用。
这种相互作用的比较有这么一个趋势,疾病的肠道菌群里边物种之间的相互作用似乎正相关的要比对照组的正相关的要多,而反过来健康的肠道菌群里面要比疾病的负相关要多。

我们把正相关(具有竞争关系的)和负相关(具有合作关系的)的参数进一步的统计,可以看到非常有意思的结果。
不管是在 genus(属)的水平、species(种)的水平,还是 strains(株)的水平,相互作用跟具体的哪一类疾病不相关,跟具体的种群也不相关,而是得到这么一个非常漂亮的、非常普适的一个对照,也就是更多的合作关系促进了健康肠道菌群的形成,而更多的竞争关系促使了疾病相关肠道菌群的形成。
我把这个规律总结为“幸福的状态都是和谐产生的,而不幸的状态都是对抗的增加导致的”。

进一步的,用 FTU 去代表一个群落之后,我们可以构建一个描述一个群落的数学模型,把它定义为建立微生物群落的一个方程。这个方程基于宏基因组全部的 FTU,以及环境因子信息和 16S rRNA 的数据,是一个微生物种群动力学的数学模型。
数学模型的一个主要的特点在于我们运用功能分类单元(FTU)去作为一个矩阵变量,把环境因子、群落结构的相关信息作为方程另外的一个条件,它就构成一个动力学的关系。

这是一组微元方程,这个模型可以很好的用在给定环境因子的条件下,进行微生物群落物种组分结构的预测。同时我们也可以通过优化计算得到一组基因,我们把这组基因定义为对构成这么一个群落的至关重要的一组功能基因,叫做 CSS 基因。
我们用肠道菌群做例子,大概能够得到 1000 多个 CSS 基因,这跟微生物基因组通常的 1000 多个叫 essential(关键)基因相比的话,它们是有差异的,不仅仅是数量上。
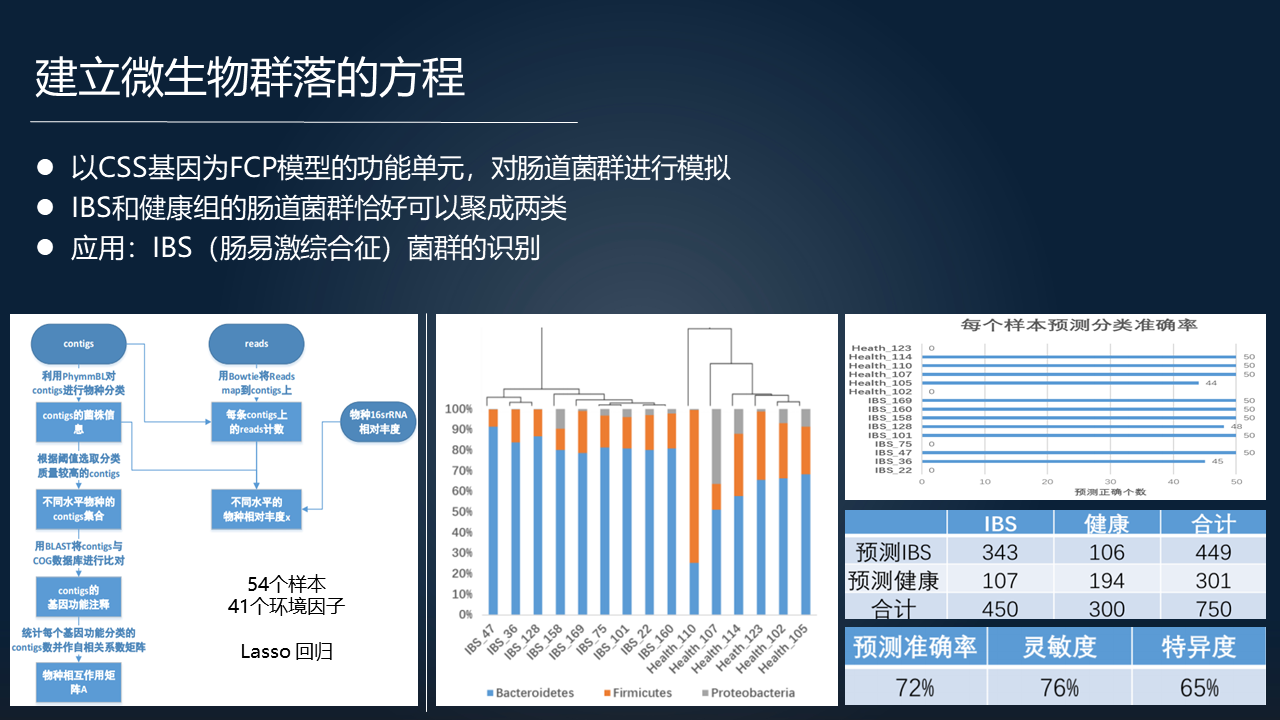
以这个为基础,我们可以得到一个应用,比如说我们用 IBS 和健康组的肠道菌群的动力学的方程去模拟,收敛以后得到 IBS 和健康的菌群可以很好的聚成两类。这启发我们未来或许可以应用于 IBS 菌群的一个临床诊断识别上。

再进一步的一个思考,就是一个菌群形成以后,当环境在变化时它是怎么去改变的?这是我们最后的一个小故事。
肠道菌群是伴随着我们成长,伴随着我们变老的。我们用 368 个人体肠道菌群的 16S rRNA 的数据,涵盖了从婴儿到 100 岁以上的年龄段,用无监督机器学习的算法去进行一个模拟归类,那么看到在没有宿主年龄信息的情况下,这些样本非常自然的按照年龄归类了。
儿童一类,成年人一类,老年人以及长寿的老人一类。随着年龄的增长,这里边的菌株或者说哪些菌属的变化值得我们关注呢?
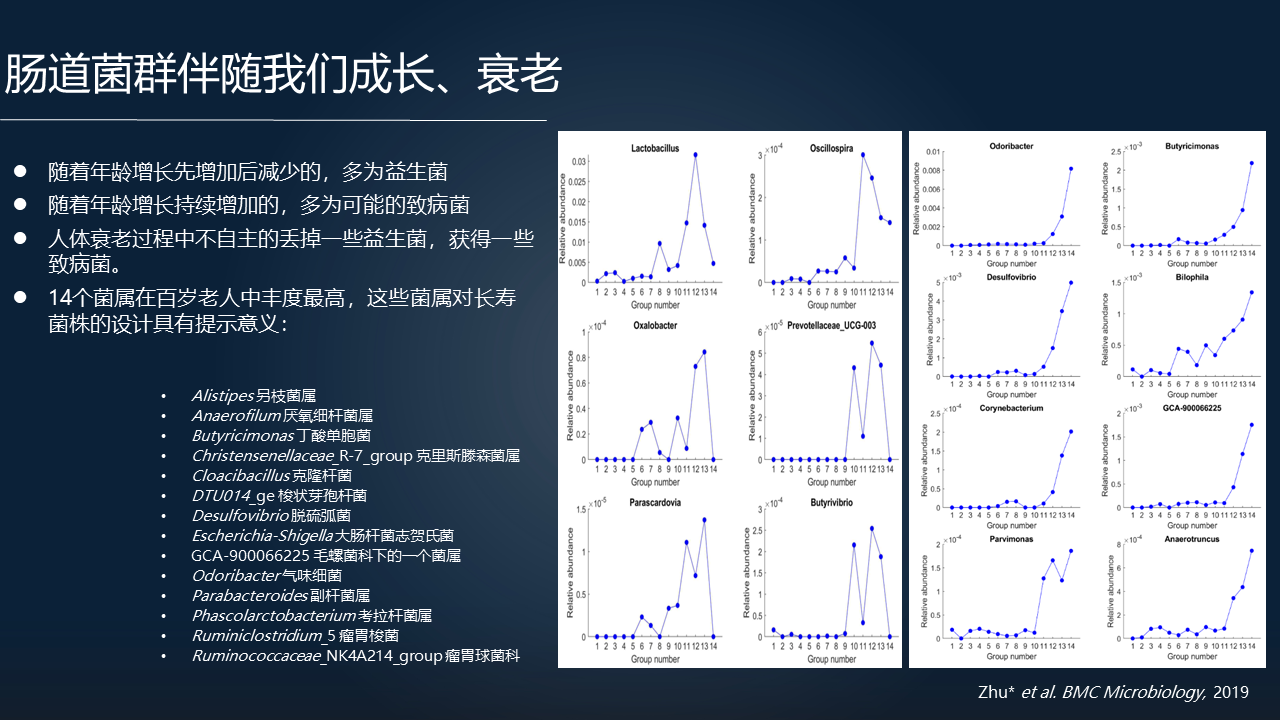
先增加后减少的可能大部分是益生菌,随着年龄增长减少的可能是跟我们衰老,与我们身体机能衰减相关的致病菌。在衰老的过程中,我们可能是在不自主的丢掉一些益生菌,并获得一些致病菌。
当然我们也能看到 100 岁以上的样本中,我们找到了 14 个可能与长寿有关的物种,非常值得进一步的关注。

我想在谈到的复杂性的时候,非常重要一点是我们现在应该去放更多的精力和更加关注的肠道菌群中细菌以外的物种,除了古菌、真菌,还要关注像噬菌体和质粒这样的非染色体 DNA 的存在。它们同时对群落的本身也是至关重要的。

总结一下我今天谈的信息,作为一种典型的复杂的微生物群落,肠道菌群的研究是非常需要生物信息学的工具、方法和思想的。
今天谈的肠道菌群的复杂性,从微生物组的水平来看的话,既可以看到菌群结构的多样性,又可以看到相关疾病的或者说环境宿主的多样性,但是我们更重视的是肠道菌群在功能上体现出更多的统一性。
一套重要的功能的单元,一套重要的功能基因的标准组成,可能是构成一个群落的关键。
未来我们应该更加的关注复杂的有机的一个微生物群落的整体,它包括细菌、古菌、真核生物以及病毒、噬菌体和质粒等等。
展望我们正在面临的微生物组学的研究,肠道菌群的研究我们需要更深入的去理解如何维持,以及更精确的去调节肠道菌群。这种调节不仅仅是从益生菌某一个菌株去调节,而是能够从精确的功能单元上去调节。
我们也可能借助合成生物学的思想途径去推动合成微生物组的研究方向。

同时我也相信,我们肠道菌群生物信息学方法和技术也是有越来越多的需求,越来越多的挑战。

谢谢大家,感谢我们课题组以及我合作的各位专家,也感谢给我们提供研究支持的平台!

谢谢大家!







