


大家好,我是宁康,来自于华中科技大学。今天非常感谢热心肠研究院提供这次机会,给大家分享一下我们在AI赋能的微生物组大数据挖掘方面的一些工作。

众所周知,微生物组学的研究目前如火如荼,在医学、生物学甚至是环境学,被应用到了非常广泛的研究领域之内。
同时科研人员也有一定的困惑,面对如此多的数据——如此多的测序数据以及其他的数据,我们如何去进行分析?
那么这个事情,首先要从数据说起。微生物组大数据,首先是属于大数据的。
对于大数据和数据相关的问题,Watson 曾经有一句很著名的话,“碱基排列的精确序列就是携带遗传信息的密码”。
同时著名的生物学家 Sulston 也说过,“生命是数字的”。
那么对于微生物组学的数据,我们认为,即使微生物组不完全是序列的,微生物组也一定是数字的。

微生物组的大数据存在各个角度的复杂性。如图所示,我们可以看到,微生物组的数据在空间上、时间上,以及包括母婴交互的这种交互性上,还有包括多组学上,均存在着不同角度的复杂性。
因此,要解析如此复杂的微生物组大数据,我们必须要通过高性能、高质量的数据挖掘方法来完成它。其中一个方兴未艾的方法,就是AI赋能的微生物组大数据挖掘。

在此我举几个例子,包括人工智能发掘微生物组数据的特征,挖掘重要的功能基因,解构时空动态变化的模式,以及通过人工智能基于微生物组的数据预测表型等等各种各样的应用。

那么,我们实验室在数据的挖掘和应用方面,也进行了一些探索。

首先给大家介绍的是,我们在组学应用上对两个重要瓶颈的一些研究。
其中,第一个瓶颈是功能基因的挖掘,主要服务于药物设计;另外一个瓶颈是研究微生物组动态变化规律,主要服务于精准医疗。
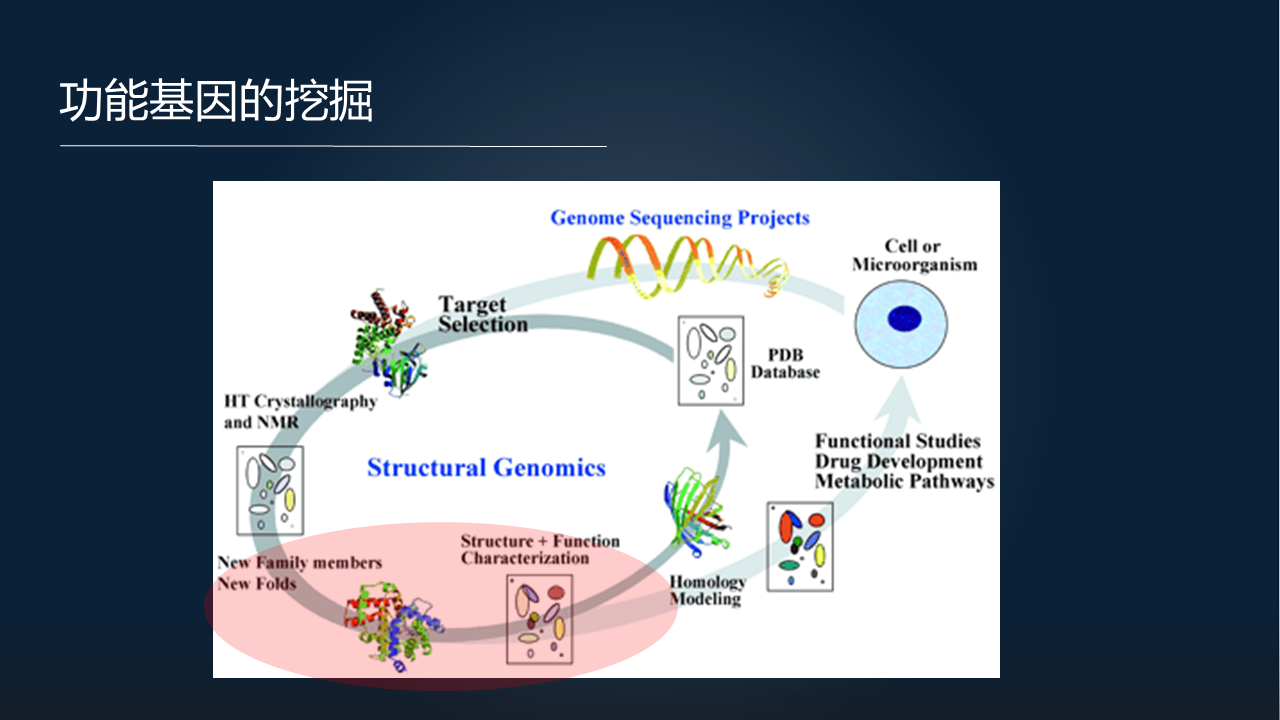
我们先讲第一个瓶颈问题。
在我拿到一个新的物种或者一个细胞之后,通常要进行一系列的分析,包括基因组的分析、功能基因的注释、重要的蛋白结构的解析,然后再回到功能验证上去。
在整个的流程中,我们现在最关心的就是,如何利用计算的方法来解构重要基因的功能以及它们的结构。

在 2017 年的 Nature Biotechnology 上面,DOE JGI的一个工作已经提示我们,仅仅是在一次过的项目里边,我们就可以在细菌与古菌的基因组里,发掘超过 50 万个新蛋白质家族,将现有类型的菌株的数目增加一倍,并将它们的系统发育多样性扩大 25%,这是非常惊人的。
同时,它提示我们,目前微生物组数据里边蕴含了大量未知的并且功能非常重要的一些资源,还有待我们去发掘。
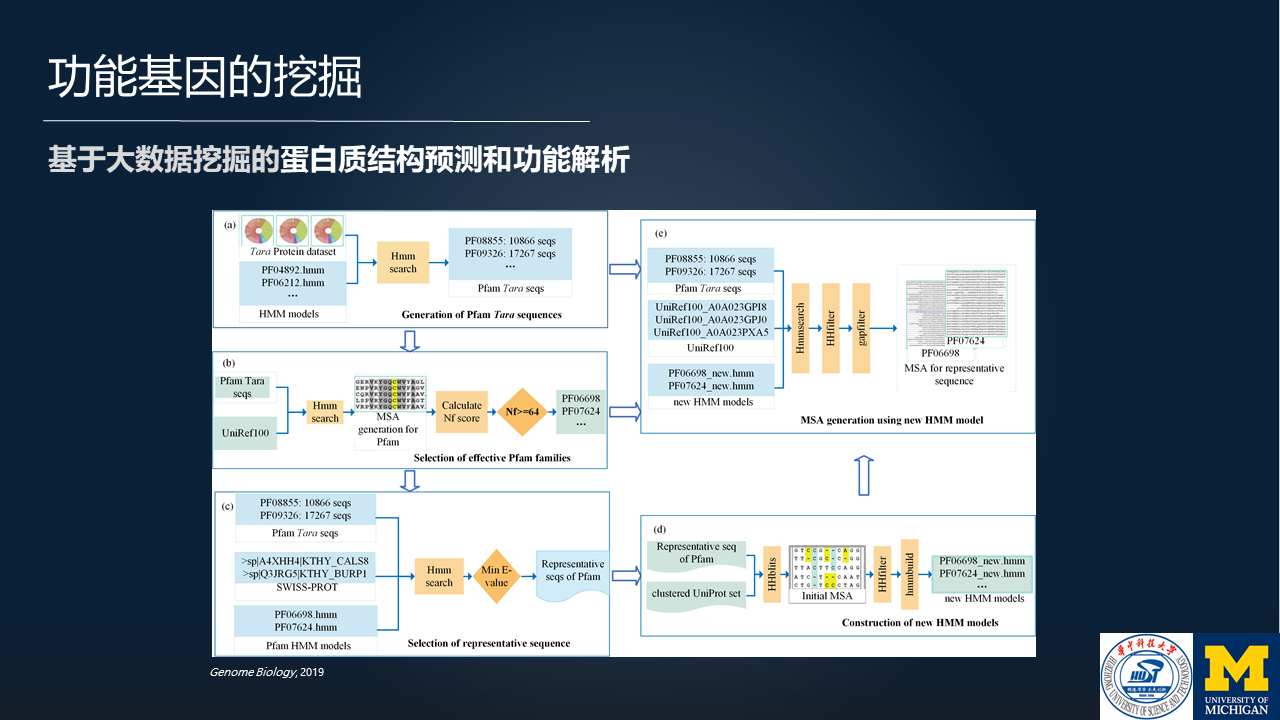
在 2019 年,我们和密歇根大学合作,针对微生物组的数据进行了一个挖掘的工作,服务于蛋白质结构的预测和功能解析。
大体的分析流程是这样的,通过微生物组学数据的分析为蛋白质家族提供同源序列,然后通过大量的同源序列的挖掘,来服务于微生物组的蛋白质结构的预测和功能解析。

主要的结果是这样的,我们基于大概 2TB 以上的微生物组的测序数据,获得了超过 300 个蛋白质家族的结构预测的结果和功能解析的结果,其中 27 个蛋白质家族的结构预测和功能解析是全新的。
如图所示,在这个图里面的一些蛋白质结构以前是未知的。其实,这并不奇怪,因为我们用了更多的数据,然后解析出来更多的蛋白质家族的结构和功能。
但是我们觉得非常有趣的一个地方在于,通过这种数据分析,我们可以知道这些新的蛋白质家族和相关的功能到底来自于哪些物种,以及这些物种到底是在哪一些环境里边富集。
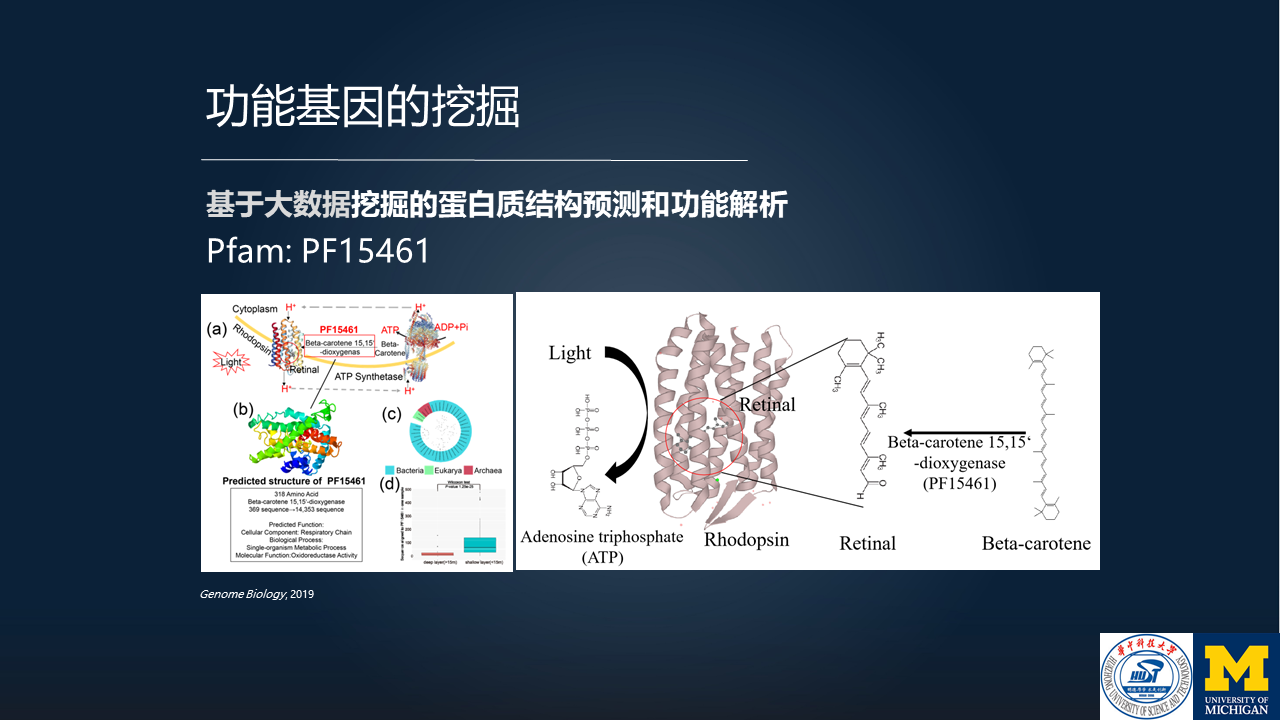
比如说下面这个例子,我们挖掘到的、预测到的 PF15461 这个蛋白质家族。
我们解析出来的结构,如这个图里所示。在这个结构里边,它有一个重要的结构域(domain),这个结构域实际上是与视紫质的光合作用有关。
我们进一步来看这个蛋白质家族的来源序列,在微生物组领域里面,它到底来源于哪个环境呢?
实际上,它是来自于海洋的生态环境。这个蛋白质所对应的物种,因为是在海洋里生存,要进行光合作用,因此它具备了这样一个功能。
那么,为什么之前的研究没有预测出来这个蛋白质家族的结构和功能呢?因为之前大量的工作是利用了公开数据集里边的人体肠道的数据,在那种环境下,这种蛋白可能并不存在,或者即使存在,信号也非常微弱。
所以,这是我们工作的主要的结论之一。
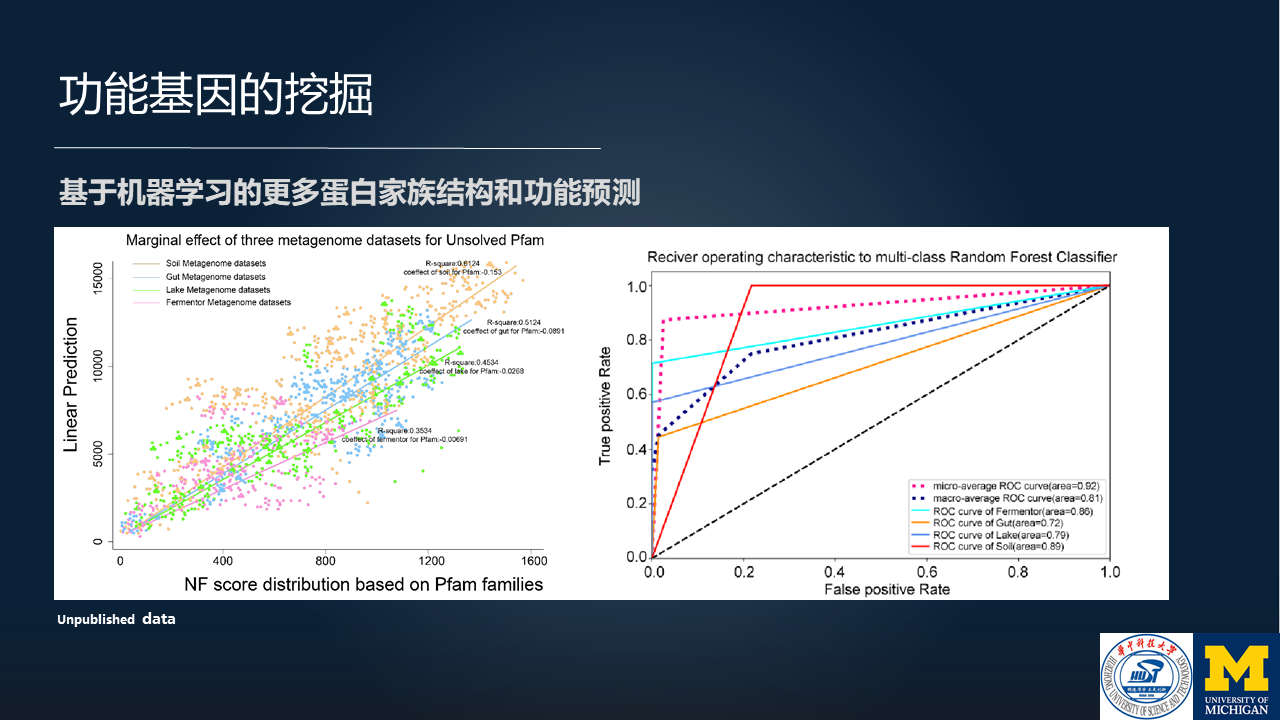
后续的话,我们最近一年来基于上述的一个认识,也就是说,在特定的环境中有特定的微生物,然后这些特定的微生物拥有特定的功能基因,因此这些微生物才能在特定的环境中生活得很好。
基于这样的一个认识,我们正在构建基于机器学习的蛋白质结构功能和预测的方法,预测结果也是非常不错的。

我们上述的基于微生物组数据的功能基因的挖掘工作,有以下潜在的应用途径:可以发掘大量未知功能基因,包括抗生素抗性基因、生物合成基因簇等等;为合成生物学提供模块;更重要的是,我们发掘的功能基因可以服务于药物发掘和药物设计。
例如,我们可以利用环境菌群的功能基因及其相关的代谢产物,来服务于新的药物发掘。

我们实验室在微生物组学和应用上力图突破的另外一个瓶颈,是微生物组动态变化规律的发掘,主要服务于精准医疗。

大家可能有这样的一个体会,每当我们到一个新的地方去之后,我们的肠道有一些不舒服的情况,这在传统的认知里边,被认为是一种水土不服的表现。
那么,水土不服在菌群层面的动态变化,到底符合什么样的规律呢?

其实之前的研究已经报道了,在两种极端的情况下,它的规律的变化情况。
第一,如果是短期的饮食变化,比如说,我短期出差到北美,那么饮食的变化对肠道微生物群落的影响,其实是不大的。
另外一个极端,如果是长期的,通常是超过一年,饮食变化也会对我们的微生物菌群产生影响,但是这种影响就是非常大了,在一定程度上会改变我们的肠型。
但是,如果我只是在一个中等范围的时间内进行饮食或者环境的变化,比如说两个月到一年这个范围内,那么对我们人体的微生物群落会有什么影响呢?目前这种情况还是不清楚的。

我们就招募了一队志愿者,看他们在半年以内的菌群变化,力图揭示水土不服的变化规律以及它们遵循的模式。
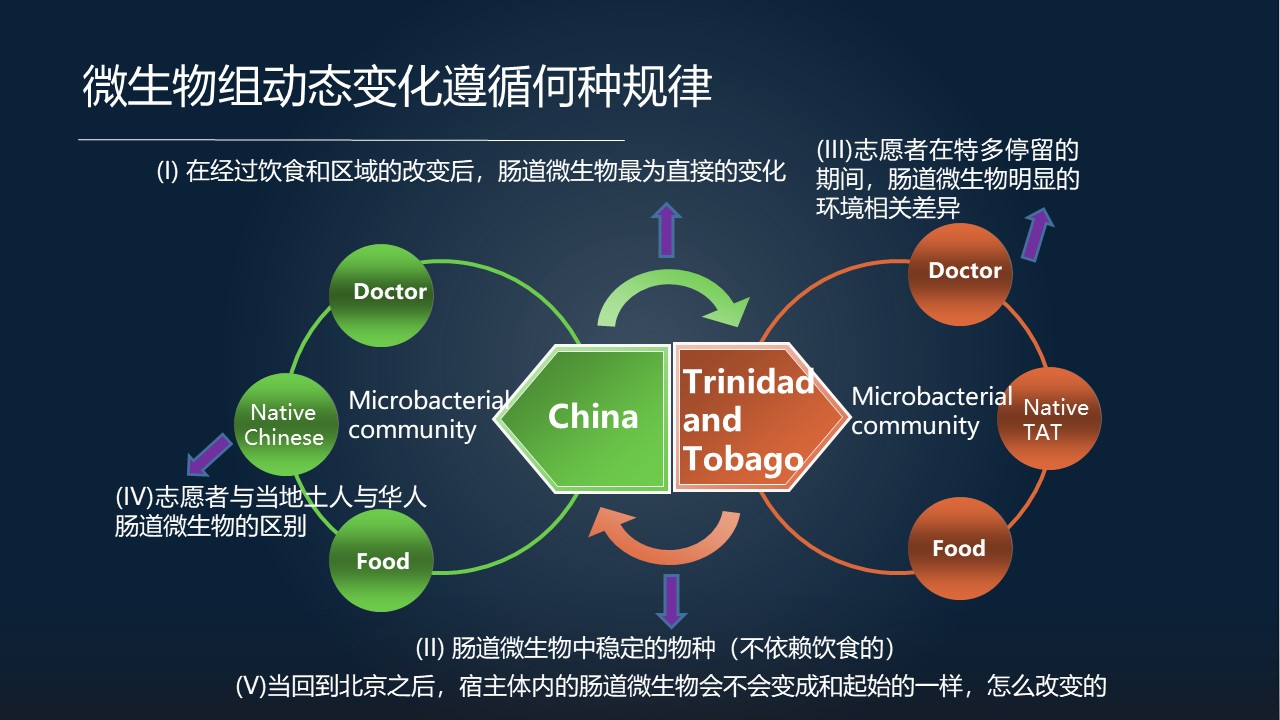
在这个研究中间,我们所想要回答的问题是,在经过饮食和区域的改变后,肠道微生物最为直接的变化是什么?这是第一个。
第二个,在这些肠道菌群剧烈的变化的过程中,有哪些物种、哪些菌,它实际上是不变的?
第三,志愿者到目的地之后,他在停留的期间,肠道菌群中有哪些细菌和环境因素高度相关?
第四,志愿者与当地的土著以及华人的肠道微生物是否有区别?这些区别是什么?
第五,当这些志愿者回到他们的出发地,也就是北京之后,宿主体内的肠道微生物会不会变成和起始一样,以及是怎么改变的?

这个是整体的样品采样的流程图,大致是从 2015 年到 2016 年,时间的间隔在一年左右,然后我们采集了整个志愿者的样本,同时我们采集了北京地区的同期样本,以及当地的土著和华人的样本。

这是 β 多样性上的一个显示图,大家可以看出来,这些志愿者的肠道菌群多样性,实际上在整个时间轴上是有一个波动的。
菌群的变化在这里看得不清楚,我们看下一个分析。

我们利用临床上常用的厚壁菌和拟杆菌之间的比例,来看一下这批志愿者肠道菌群的变化。
左下图,我们看到了从 T1 一直到 T6 的时刻,它们呈现一个很明显的这样的波动的情况。

那么这种波动的情况到底符合一个什么规律呢?我们通过 PCoA 的这种分析方法。
首先把两边的对照,一边是北京的正常人,一边是当地土著的正常人,一个用红色表示,一个用蓝色表示,放在这里。
我们跟着折线的规律往下走,当我们把志愿者的样本用紫色表示,往这个图上面进行映射的话,我们发现紫色的样本一开始是跟红色的样本很接近的,然后慢慢地聚集到这些下面的蓝色样本,也就是当地的土著上面去。
当他们回来之后,我们用绿色的点在图中表示,我们发现它们又恢复到原来北京的正常人的菌群上面去了。
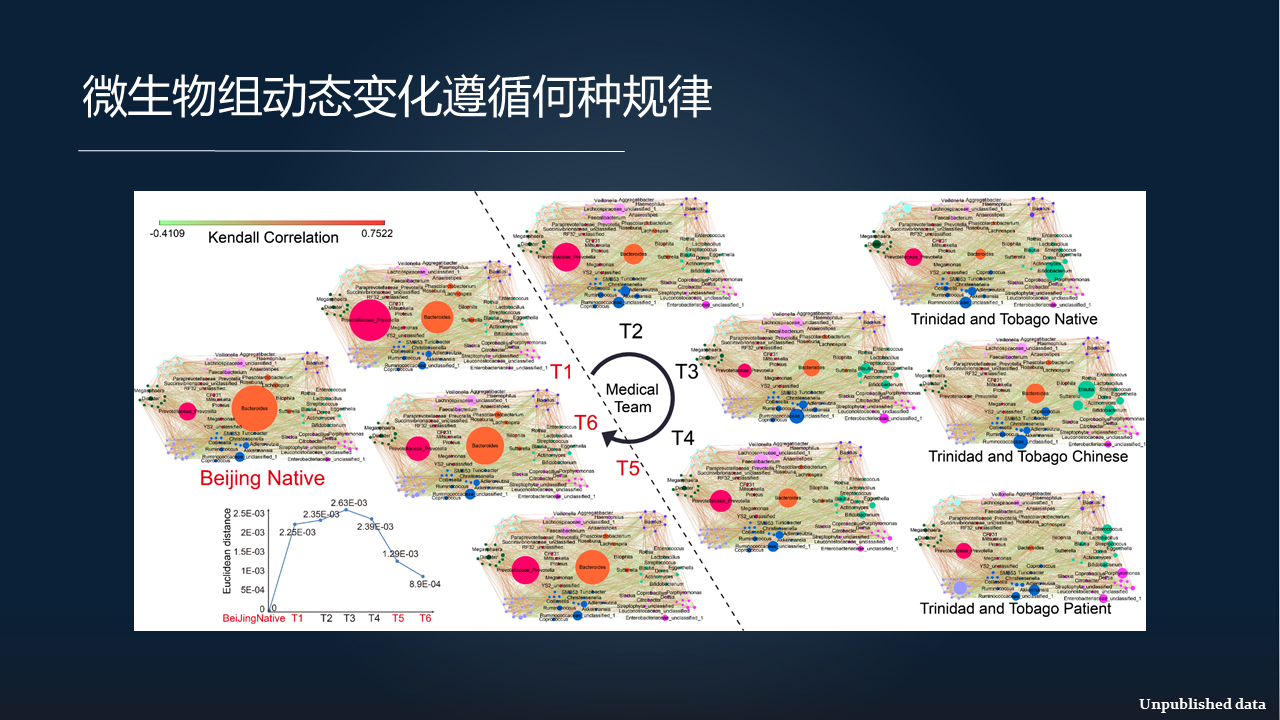
我们在这里又用一个网络的形式,来把这个规律再阐述一遍。
左边是北京的正常人,右边是当地的土著,我们把它分别作为正对照和负对照,那么一个虚线划开了这些样本到底是采自于北京,还是来自于目的地。
在 T1 时刻,也就是出国之前,他们的肠道菌群是跟北京的正常人是很相似的。
到了目的国之后,他们的这些肠道菌群马上就会变得跟当地的土著很相近。同时,这些相似程度随着时间的推移,会变得越来越像。
但是当他们回到中国之后,他们的菌群又马上变回到更接近于北京的正常人。实际上是这样一个规律。

那么,这种动态变化规律在细菌的单个菌株上面也同时存在。

另外我们想回答的一个问题是,这种动态变化规律实际上和哪些饮食因素有关呢?
从这个图里面我们可以看出来,它实际上和目的国的西式饮食里边的高糖、高脂以及奶制品等等这样的饮食特征,是高度相关的。

因此,我们在这个工作中,在菌群的层面阐述了“水土不服”的动态变化规律,揭示出了肠道微生物与环境和饮食有着密切关系,确定了这种动态变化的模式。
同时,我们通过系统性地采样和数据分析,揭示了“菌群适应性和恢复性”的模式,发掘了肠道菌群的“强韧性”的特征。
这个工作是我们和中科院、北京计算所以及北京世纪坛医院一起合作完成的。

那么这个工作在临床上、精准医学上有什么应用呢?至少有两个提示。
第一个提示是,菌群的强韧性会潜在地减弱抗生素等药物的药效。也就是说,大家在服用了抗生素等其他药物之后,如果不相应地改变饮食习惯或者改变饮食的内容,那么它的药效有可能会被减弱。
另外一点,对健康菌群的长期跟踪具有必要性。因为即使是半年以上的饮食和环境的这种改变,实际上菌群的恢复也是会很快的。

这个工作还在其他的哪些领域有所应用呢?
在这里我列出了几个比较典型的应用,包括援外医疗、候鸟老人、援疆援藏以及远程投送。
上面介绍了我们在两个瓶颈问题上的一些探索,那么接下来可以给大家分享一下,我们在微生物组大数据挖掘方面的一些想法,就是微生物组大数据以后可以往哪个方向进行挖掘。
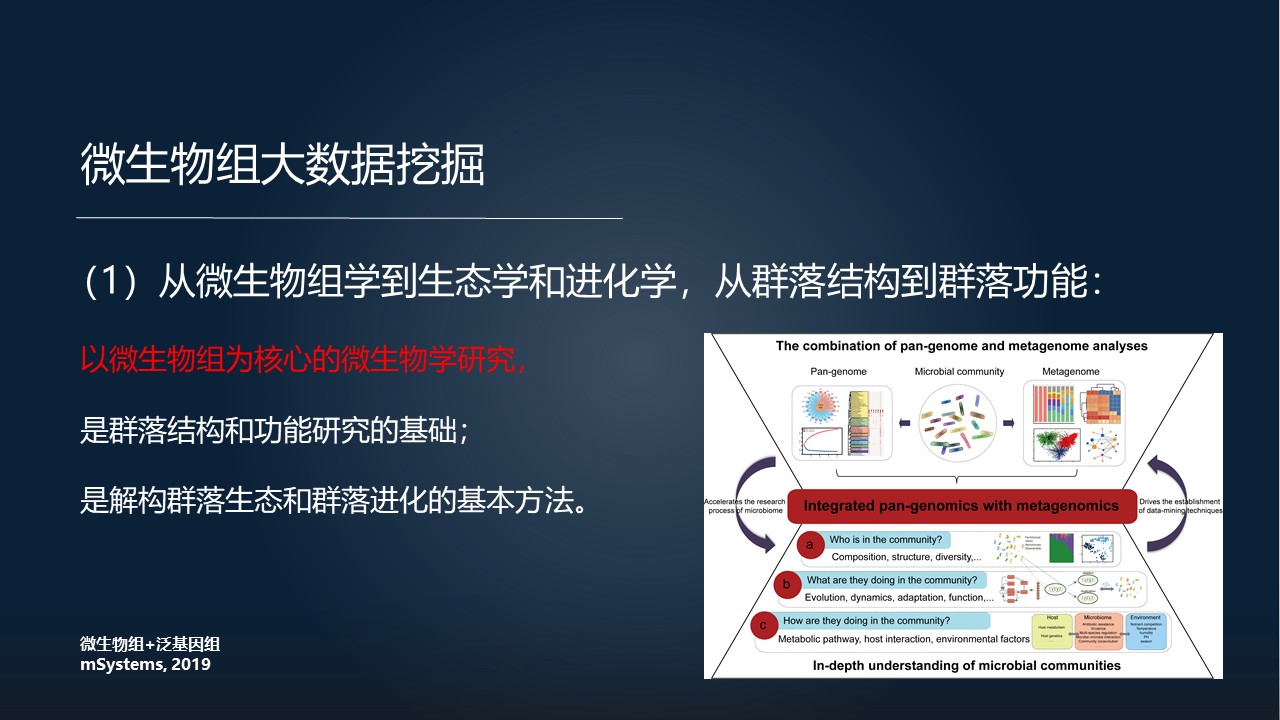
第一个,从微生物组学到生态学和进化学,从群落结构到群落功能。
我们认为,以微生物组为核心的微生物组学研究,是群落结构和功能研究的基础,是解构群落微生态和群落进化的基本方法。
如果将微生物组学和泛基因组学,以及其他的微生物相关的生态学和进化学相结合在一起的话,将会更深层次地解构微生物群落以及单个的微生物。

第二个方面,我们认为微生物组学的研究已经从小数据发展到了大数据。在面对这个问题,我们必须要基于高性能的计算,来进行微生物组大数据的挖掘。
在这里给大家列出来的,是我们在今年发表的一篇文章,相关的工作实际上已经证明,我们可以针对100万个或者以上的微生物组样本,在非常短的时间内进行全局地搜索。这种样本的搜索,可以有利于我们更好地进行微生物组的规律挖掘以及模型构建。
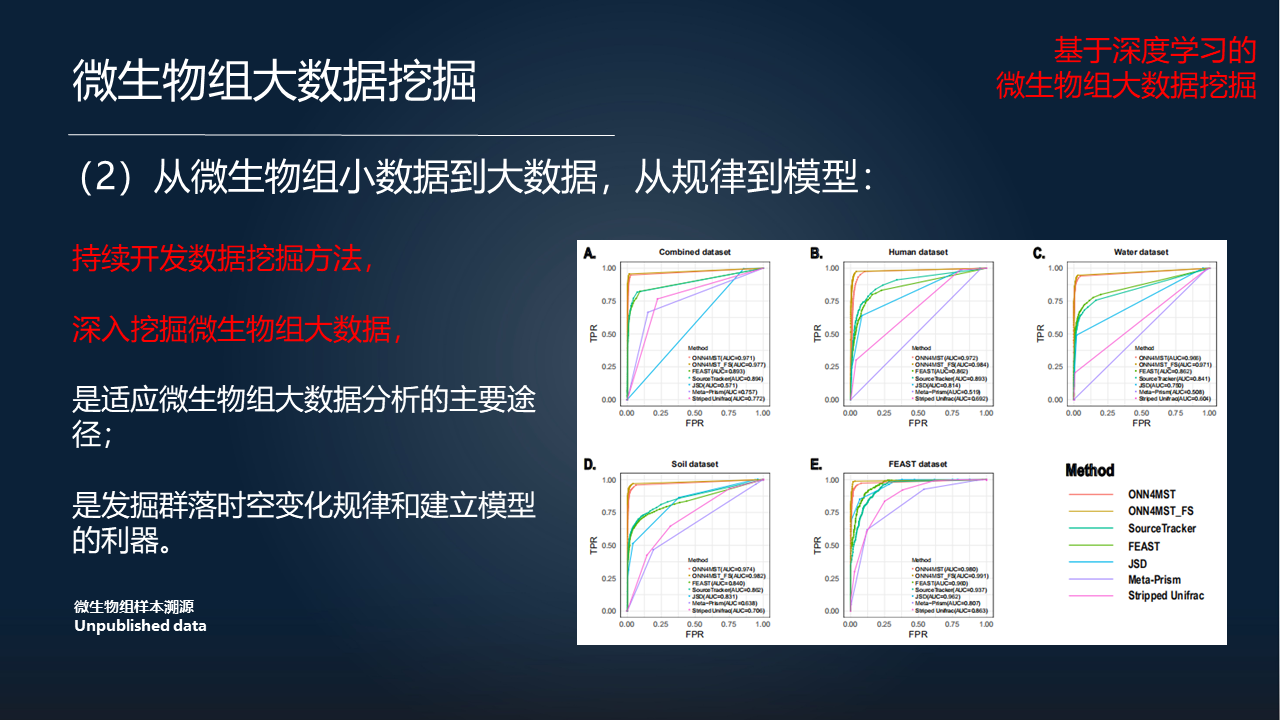
所以在这里我们认为,持续地开发数据挖掘方法,深入地挖掘微生物组大数据,是适应微生物组大数据分析的主要途径,是发掘微生物群落时空变化等等其他一些变化模式以及建立相关模型的利器。
例如,这个图里展示了我们在微生物组样本溯源上的一些探索。如果我们用到了超过100万个微生物组样本的话,我们在微生物组样本的溯源上,其实将会达到前所未有的准确性,以及前所未有的效率。

第三个方面,从微生物组到多组学,从相关性到因果性。
我们认为以微生物组为核心的多组学研究,配合基于大数据的相关性和因果性的推断方法,是深入解构群落发生发展规律的重要手段。
例如在这个例子里边,这是我们在今年发表的一篇关于运动员的微生物组、代谢组与表型组的整合研究。
在其中我们发现了,对于高水平运动员来说,他们体内的肠道微生物实际上是连接饮食、生活习惯、体检指标以及最后的运动成绩的非常重要的一个纽带。

第四个方面,从微生物组数据挖掘到实验设计的全程参与。我们认为,从实验设计开始贯彻微生物组大数据的思维,是完成高质量的微生物组研究的必经之路。
这是我们和北京协和医院重症监护中心的合作,在临床微生物组研究方面的特征挖掘。通过前期的参与实验设计,到中期的样本搜集、测序数据的获得以及数据的分析,到后期的通过结果一定程度地进行预测和干预。
我们觉得这一套思路可能今后将变得越来越重要。

最后总结一下微生物组大数据。
我们通过刚才的几个例子已经知道了,微生物组是数字的,微生物组不只是序列的。因为我们还知道,微生物组还会包含生物图像以及结构生物学等等其他的组学数据,或者是非组学的图像数据。
但是不管什么数据,AI 赋能的数据挖掘将会变得越来越重要。

在这里,我们已经介绍到了临床方法、数据挖掘方法、生物学的验证方法。大家可能会觉得,如此多的门类领域如何通过一个实验室来完成?
其实我们想要强调的是,从群落样本到微生物组的大数据,从微生物组大数据挖掘
到相关应用,解决所有这种跨领域的交叉研究问题的核心方法,是合作研究。
今后的微生物组学研究,一定是基于更大范围、更深入的合作才能够完成的。

最后非常感谢我们实验室的成员,感谢我们世界各地的合作者。
谢谢大家。






